


镍基高温合金因具有优异的高温性能而被广泛应用于航空航天发动机用涡轮盘及叶片等的热端部件[1~3]。涡轮叶片在重复运行周期期间,应力和温度会发生显著的波动[4]。为了能更好地适应叶片恶劣的高温服役环境,衍生了热障涂层(TBC)技术[5]以提高涡轮前进口温度。热障涂层是由低热导率陶瓷面层和金属黏结层组成的隔热系统,主要有双层结构、梯度结构和多层结构[6,7],最为典型的是双层结构,其中的金属粘结层一般为MCrAlY或Pt-Al合金[8]。改善涂层各界面的热匹配性,可以提高热障涂层的抗高温氧化和高温腐蚀能力。由于叶片工艺条件的局限性,S元素不可避免地残留在合金中,研究[9]发现10-6量级的S元素含量就足以对合金性能产生严重的影响。Walsh和Anderson[10]通过Auger电子能谱(AES)发现S元素主要偏聚在高温合金中的晶界处;Dong等[11]也通过此方法研究了S元素在IN718高温合金中的偏析现象,发现其主要在碳化物界面处偏聚。服役时在覆有涂层的高温合金叶片中,S元素倾向于在金属基体/涂层界面处富集[12~14]。国内外学者通过不同角度及尺度计算方法对S元素在高温合金与涂层界面的情况也开展了系列研究。Ozfidan等[15]运用第一性原理计算方法研究了S元素对NiAl(110)/Al2O3(0001)界面结构和结合强度的影响规律,并通过研究S元素偏聚于金属基体/涂层界面的现象解释了界面结合强度降低的原因。研究指出,S元素易偏聚在NiAl涂层一侧,虽然导致最近邻区域AlNiAl—O键长减小,但是AlNiAl—O和Ni—AlOxide键数量减少的影响大于其键长减小的影响。镍基高温合金中有序L12结构的γ'-Ni3Al相嵌入无序fcc结构的γ基体,为体系提供了强化的条件,大大提高了镍基高温合金的热机械性能[16],因此对S元素在两相界面处影响的研究也尤为重要。Chen等[17]研究了S在镍基单晶高温合金γ/γ'相界处产生的脆化效应,发现S原子和界面处的Ni原子形成很强的键合,从而提高了合金的剪切强度,同时剪切强度与内聚强度的比值也随之增大,于是产生了界面脆化。Fedorova等[18]采用第一性原理计算方法,建立了2种计算黏附功模型,发现随着S含量增加,氧化膜与合金基体间的黏附性降低;Nychka等[19]认为S的存在降低了Al2O3与金属之间的键能,进而损害了氧化物与界面的断裂韧性。考虑到以上S元素造成的不利影响,亟需对S元素偏聚的影响机制开展研究,为界面设计提供参考数据和研究基础。同时,由于难以通过实验全面获得S元素对合金微观影响的潜在机制,故本工作采用第一性原理的方法开展系列计算。
本工作应用第一性原理计算方法同时研究了镍基高温合金/涂层(NiAl)以及NiAl涂层/氧化层(Al2O3) 2种界面体系。通过构建相应的界面(无S/含S)模型,再采用单点能量计算来确定相界面间的最佳距离,以两相间距d为自变量、计算得到的体系能量E为因变量,绘制E-d曲线,以确定最优构型。计算2种体系的界面黏附功及偏析能以验证含S界面的偏聚行为。进一步以S的偏聚现象为切入点,对无S/含S界面体系附近原子进行了电子结构分析,阐明S元素偏聚及界面黏附功数值变化的机制,进而揭示含S界面体系的微观影响机理。本工作旨在突破实验方法难以达到的对含S界面电子结构微观尺度研究的难题,为实际应用中改善先进叶片性能提供计算数据,并为镍基高温合金叶片材料及涂层的制备领域提供理论参考。
1 计算方法
1.1 模型构建与计算过程
在密度泛函理论(density functional theory,DFT)基础上,本工作计算基于DFT的VASP (Vienna ab initio simulation package)软件[20],采用投影增强波(projector augmented wave,PAW)法[21]描述元素的离子和电子的相互作用,交换关联泛函采用广义梯度近似(generalized gradient approximation,GGA)的PBE (Perdew-Burke-Emzerhof)泛函[22]。为了保证DFT计算的准确性,对Ni3Al/NiAl、NiAl/Al2O3的无S/含S的界面结构模型进行了平面波能量截断能为400 eV和k点网格为4 × 4 × 1的Monkorst-Park方案的优化。对于Ni3Al/NiAl体系的计算模型,电子自洽与离子弛豫的收敛标准为10-5 eV和-2 × 10-1 eV/nm;对于NiAl/Al2O3体系的计算模型,上述取值分别为10-4 eV和-2 × 10-1 eV/nm。真空层距离设置为1 nm,在结构优化时进行仅固定体积的弛豫,固定端部的原子,对界面附近的原子进行弛豫。以上计算条件为无压力状态下的恒定温度(0 K)。建立模型过程中发现,对Ni(111)/NiAl(110)模型进行扩胞时总原子数过多,Ni(111)和NiAl(110) 2者的界面模型宽度匹配也存在一些差异。考虑到镍基高温合金优异的性能源于L12型有序γ'-Ni3Al相分散在fcc型无序γ基体相中(体积分数可高达70%[23])以及bcc-fcc相界面的Kurdjumov-Sachs (K-S)取向关系(即对于fcc-bcc相界面,fcc的最密排面(111)与bcc的密排面(110)可能结合成半共格界面),将Ni3Al模型替代镍基高温合金。通过获得的Ni3Al、NiAl以及Al2O3的单胞结构依次建立单相模型Ni3Al、NiAl、Al2O3;因Ni3Al-fcc、NiAl-bcc、α-Al2O3最紧密和最稳定的表面分别是(111)、(110)、(0001),进而建立相应的表面模型:Ni3Al(111)、NiAl(110)、Al2O3(0001);纯净界面模型以及含S界面模型,以表征纯净界面与S元素存在时界面附近发生的变化。据报道[15],NiAl/Al2O3界面体系中以Al原子为终端的Al2O3(0001)表面比以O原子为终端的表面更稳定。因此,将所建立的模型中NiAl/Al2O3界面部分减掉一个O原子,以基于这一模型展开计算。在建立含S界面模型时,参考Carling和Carter[24]研究中S元素稳定在NiAl(110)的2Ni-Al三重位点位置(吸附能最低)的结果,虽然界面体系中有多个这样的位点,但是选择S加入到尽可能少地破坏界面上Ni—Al键的位置;并指出这种类似的初始结构中,Hf原子会向更有利的位置大幅度移动。因此对于本工作含S元素的界面结构来说,体系中有一定的空间使相对Hf原子较小的S原子重新调整位置,经过结构优化,可以保证S原子加入后界面结构的相对稳定。
将用于微观组织观察的样品(样品基底的化学成分(质量分数,%)为:Cr 19.24,Co 19.18,W 6.01,Al 1.98,Ti 3.76,Ta 1.55,C 0.19,Nb 1.13,Ni余量)机械研磨抛光后,采用4 g CuSO4 + 20 mL C2H5OH + 20 mL HCl的侵蚀剂浸泡3~8 min,使用Imager M2m光学显微镜(OM)观察金相组织。将样品重新研磨抛光后,使用20 mL浓H2SO4 + 80 mL CH3OH的电抛液,在15 V电压下电抛5 s,在150 mL H3PO4 + 10 mL浓H2SO4 + 15 g CrO3的电解液中电解3 s,使用Supra55场发射扫描电镜(SEM)观察截面微观结构。根据具有热障涂层的镍基高温合金截面显微组织(图1a),将所绘制的合金基体-黏结层-热障涂层示意图作为在第一性原理计算建模阶段的参考(图1b),首先选取Ni3Al建立单相模型,进而构建计算模型:即Ni3Al为基体,其上依次为NiAl涂层、Al2O3氧化层。
图1

图1
具有热障涂层的镍基高温合金截面显微组织及合金基体-粘结层-热障涂层示意图
Fig.1
Cross section microstructure of nickel-based superalloy with thermal barrier coating (TBC) (a) and schematic of microstructure of nickel-based superalloy (b)
图2
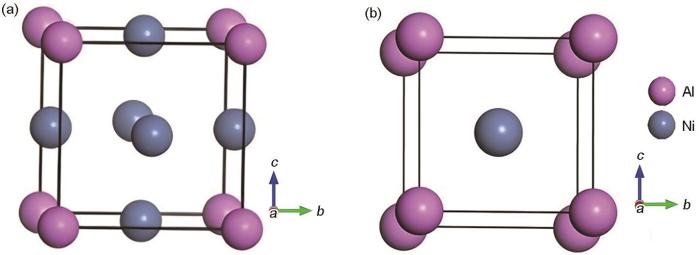
图3
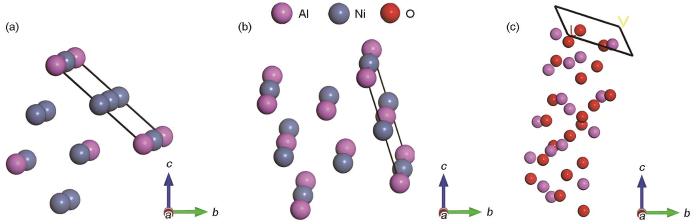
图3
Ni3Al(111)、NiAl(110)和Al2O3(0001)表面模型
Fig.3
Surface models of Ni3Al(111) (a), NiAl(110) (b), and Al2O3(0001) (c)
图4
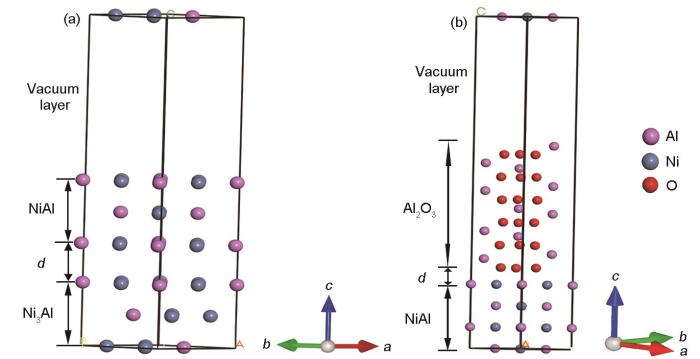
图4
Ni3Al/NiAl相界面模型和NiAl/Al2O3相界面模型
Fig.4
Phase interface models of Ni3Al/NiAl (a) and NiAl/Al2O3 (b) (d—phase distance between the upper region of Ni3Al(111)/NiAl(110) surface and the lower region of NiAl(110)/Al2O3(0001) surface)
式中,下标α、β代表2种材料,a为晶格常数,a'表示2种材料平均的晶格常数,将2者晶格常数差异定义为晶格错配度。一般来说,界面的晶格错配度小于5%时,由晶格错配所引起的能量变化可以被忽略。2个界面模型所得晶格错配度分别为0.68%和4.98%,表明建立的界面模型是在软件系统允许误差范围内的。绘制E-d图,以得到最为稳定的结构,即合理的初始构型。对于Ni3Al/NiAl相界面模型,d值范围在0.06~0.32 nm适当选取步长;对于NiAl/Al2O3相界面,d值范围在0.06~0.21 nm适当选取步长。根据计算得到的E值,缩小2种界面模型能量最低点的范围后,在此小范围内缩小步长,进行相应体系的静态计算。图5和6分别为Ni3Al/NiAl和Al2O3/NiAl 2种体系对应的E-d曲线图。2种模型的相间距在一定范围内都存在某一间距的数值使得系统总能最小,达到相对来说最为稳定的状态。Ni3Al/NiAl界面模型中最低能量对应的相间距在0.252 nm处,对应的体系总能量约为-120.772 eV。对于NiAl/Al2O3界面模型,在相间距为0.06 nm附近时,由于数值小于体系中所有原子半径最小的O原子(0.074 nm),所得数据可能存在一些偏差,故该体系最低能量对应的相间距在0.160 nm处,对应的体系总能量值约为-288.166 eV。
图5

图5
Ni3Al/NiAl界面体系能量-相间距(E-d)图
Fig.5
E-d plot of Ni3Al/NiAl interface (E—energy obtained from static calculation of the system at the corresponding phase distance d )
图6
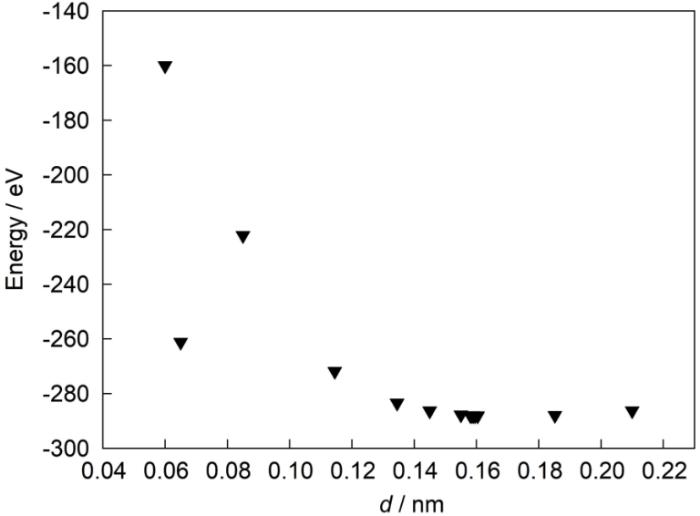
图7
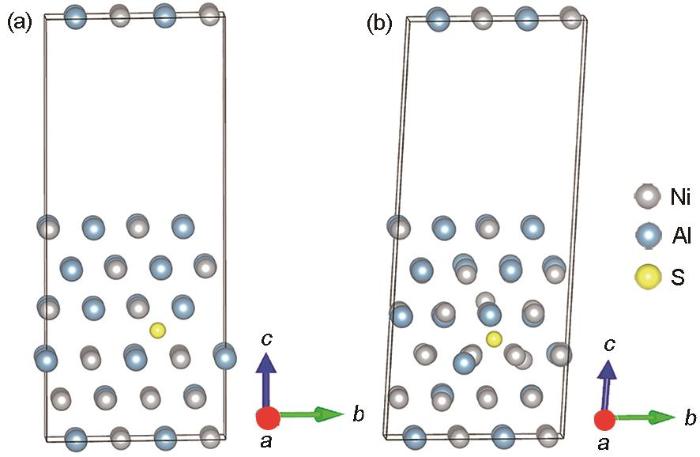
图7
含S元素Ni3Al/NiAl界面体系结构优化前及优化后的模型
Fig.7
Ni3Al/NiAl interface structure within S element (a) and the system of Ni3Al/NiAl interface after structure optimization (b)
图8
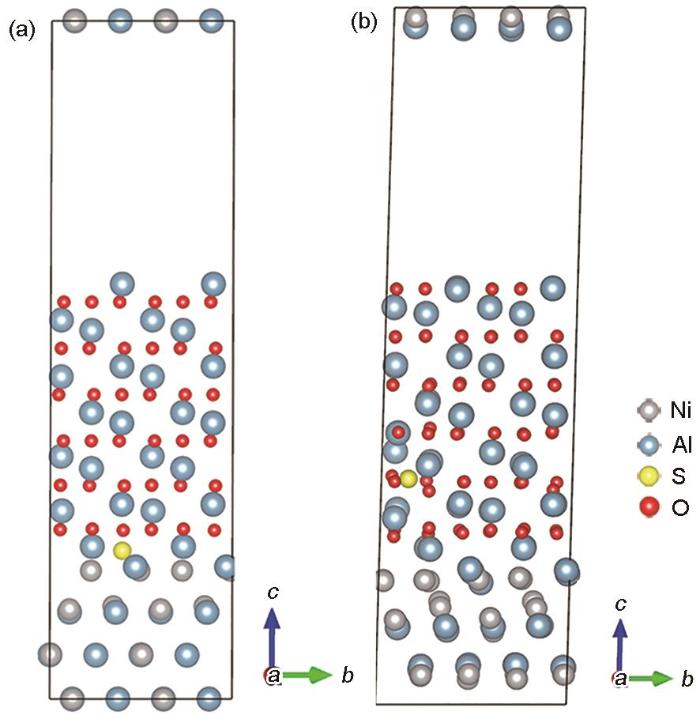
图8
含S元素NiAl/Al2O3界面体系结构优化前及优化后的模型
Fig.8
NiAl/Al2O3 interface structure within S element (a) and the system of NiAl/Al2O3 interface after structure optimization (b)
1.2 计算参量及处理方法
1.2.1 界面黏附功
界面黏附功可用来描述界面的行为及其黏接特性,是衡量界面稳定性的重要物理参量之一。它定义为在不产生塑性变形的情况下,将界面可逆地分离为2个自由表面所需的能量,由于在实际断裂实验中所需要的能量总是远大于理想的黏附功,因此认为计算所得数值是断裂实验获得的黏附功的下限[15]。黏附功可由
1.2.2 偏聚能
由界面黏附功的计算结果可知,合金中S元素的存在会附近降低界面的结合能力,因此衡量S原子偏聚趋势也尤为重要。镍基高温合金中,杂质元素向界面偏聚的趋势可以用偏聚能Eseg来衡量,定义如下[29]:
式中,X表示模型界面(in)或基体(bulk),EX 和EX - S分别表示S原子在X处取代前后的总能,
此处EX 和EX - S分别指S原子在X处掺杂前后的总能量,
式中,Einterface(doped at interface)表示掺杂原子在界面处的体系静态计算所得总能量,Ebulk(in bulk)表示掺杂原子在体相中的体系静态计算所得总能量。
1.2.3 二次差分电荷密度
当一个体系化学成分或几何构型改变之后,电荷会重新分布,描述其分布情况的参量为二次差分电荷密度,其表达式为:
式中,Δρ代表成键后与成键前电子密度的差值,ρAB表示S元素存在的界面经过优化后计算出的电荷密度,ρA、ρB分别表示纯净界面、存在元素S体系中原子的电荷密度。二次差分电荷密度图可以直观体现原子之间的相互作用的特征,达到定性研究界面处电荷转移情况的目的。通过第一性原理计算方法计算并进行数据处理得到平衡态下2种界面模型纯净界面体系和含S界面体系在三维实空间差分电荷密度的分布,为更全面地分析电荷聚集/损失的具体分布空间,绘制了二维电子密度图,得到体系中特定原子(2种界面模型中S近邻的Ni、Al、O原子)的电荷密度变化、成键能力(原子之间连接紧密程度)及成键特性的判断,进而分析界面结合强度的变化和成键本质[33]。
1.2.4 电荷定量分析
二次差分电荷密度图可以定性反应界面处电子的转移情况以及成键情况,若想定量表示界面原子之间的电子转移及分布情况,需要结合界面原子Bader电荷的计算,进一步研究原子之间电子转移情况。Bader电荷是一种将材料的总电荷分解到原子电荷的计算方法,可定量分析原子成键前后电荷的转移情况,以理解电子如何在S原子和其近邻原子之间转移以及S原子如何使体系电子转移量发生变化的。研究中主要计算界面处相关原子的Bader电荷,即选定界面两侧近邻S原子的Ni、Al原子以统计界面附近弛豫原子层总计转移电子的情况。
1.2.5 电子局域化函数(ELF)分布
ELF是一个三维空间函数,能够使电子局域性、离域性图形化表现。ELF数值范围在0~1之间,与电荷密度相比其增大了键合特性,ELF还可以描述在多电子体系中电子对的概率以及界面上原子位置的相对变化趋势。理论上,它描述了在一个电子附近找到一个自旋相同的电子的概率,这个电子是相对于某个位置的电子的。ELF值接近或大于0.75时,说明存在高度电子局域化,电子不容易远离原子核,一般形成共价键或离子键;在极端离域化时接近0,此时电子局域性很弱,电子很容易离开正离子,此时一般形成金属键类型,在价电荷分布类似于均匀自由电子气体的区域接近1/2[34]。S原子偏聚到界面处时会改变界面附近Ni、Al、O原子周围的化学键,因此为进一步判断界面处原子成键特性,本工作进一步选定晶面进行电子局域化函数分析,达到放大分析其中键合特征的目的。
1.2.6 态密度(DOS)分布
态密度可以作为能带结构的一个可视化结果,能够更直观地反映出电子在各个轨道的分布及杂化情况,有总态密度(total density of states,TDOS)、分波态密度(partial density of states,PDOS)、局域态密度(local density of states,LDOS)及局域分波态密度(local partial density of states,LPDOS)等。系统中特定元素的电子状态可以用局域分波态密度来表示,即将态密度分解到每个原子上面的s、p、d等轨道。通过绘制无S和含S体系的TDOS、S原子及其近邻的Ni、Al、O原子的LPDOS,分析特定原子中电子之间的相互作用和S元素对合金及涂层体系微观电子层面的影响。
2 计算结果与分析讨论
2.1 界面黏附功
对于无S和含S的Ni3Al/NiAl界面体系,黏附功计算结果如表1所示。对于无S界面模型,结合
表1 Ni3Al/NiAl界面模型界面黏附功计算值
Table 1
| System | A / nm2 | ES1 / eV | ES2 / eV | ES1/S2 / eV | Wad / (J·m-2) |
|---|---|---|---|---|---|
| Pure interface | 0.8903 | -235.024 | -241.350 | -494.860 | 3.427 |
| Interface with S | 0.8967 | -235.024 | -241.350 | -496.204 | 3.394 |
同理,对于无S和含S的NiAl/Al2O3界面体系,黏附功计算结果见表2。对于纯净界面模型,Wad ≈ 1.162 J/m2;对于含S界面模型,ES1/S2 ≈ -1178.733 eV,单个S原子在模型中的总能量约为-1.070 eV,故计算可得Wad ≈ 0.449 J/m2。S元素在合金/NiAl涂层中会使得界面黏附功降低约0.96%,使界面体系变得不稳定。S元素在NiAl涂层/Al2O3体系中使得界面黏附功降低了8.98%,为维持自身稳定性,具有强烈偏聚到涂层一侧的倾向。在涂层/氧化层界面处,S原子的存在导致界面黏附功相比于纯净界面下降了61.36%,此体系的界面结合强度下降的幅度较大,下文将从微观角度阐述其下降诱因。此外,由于金属-陶瓷层状复合材料的强度与界面结合强度具有相关性,即界面强度过低会导致材料在加载过程中发生基体与陶瓷之间界面的脱落[37],因此在含有热障涂层的镍基高温合金体系中,界面结合强度的评估至关重要。
表2 NiAl/Al2O3界面模型界面黏附功计算数值
Table 2
| System | A / nm2 | ES1 / eV | ES2 / eV | ES1/S2 / eV | Wad / (J·m-2) |
|---|---|---|---|---|---|
| Pure interface | 0.8325 | -883.450 | -293.219 | -1182.710 | 1.162 |
| Interface with S | 0.8330 | -883.477 | -292.060 | -1178.733 | 0.449 |
2.2 偏聚能
由结构优化前后的Ni3Al/NiAl界面体系模型及NiAl/Al2O3界面体系模型(图7和8)可以看出,S元素倾向于稳定在涂层NiAl和氧化层Al2O3中。结合计算结果中数值及
2.3 二次差分电荷密度
对于Ni3Al/NiAl界面,二次差分电荷密度结果如图9所示,图中黄色区域表示电荷密度增加,蓝色区域表示电荷密度减少。可见界面附近S原子周围发生了明显电荷转移的区域。在含S元素的Ni3Al/NiAl界面体系中,与S原子近邻的Ni附近电荷密度增加,界面附近处基体一侧和NiAl涂层一侧Ni、Al原子之间均发生了电荷转移。Al元素周围电荷密度减少,其程度不如Ni原子变化明显,S原子附近电荷密度也出现明显减少,且S原子与近邻的Ni原子之间存在较大程度的电荷转移区域。图10为含S原子体系(010)平面的电荷密度分布图,给出了界面附近S原子与其近邻的Ni、Al原子之间的电荷转移情况。可见S原子倾向于与近邻NiAl涂层中的Ni原子与之间成键,与Chen等[17]的研究结论一致。故在此界面体系中,界面附近偏聚的S原子倾向于与近邻NiAl涂层中的Ni原子之间成键,导致合金中Ni、Al原子之间局部金属键的连接紧密程度减弱。
图9
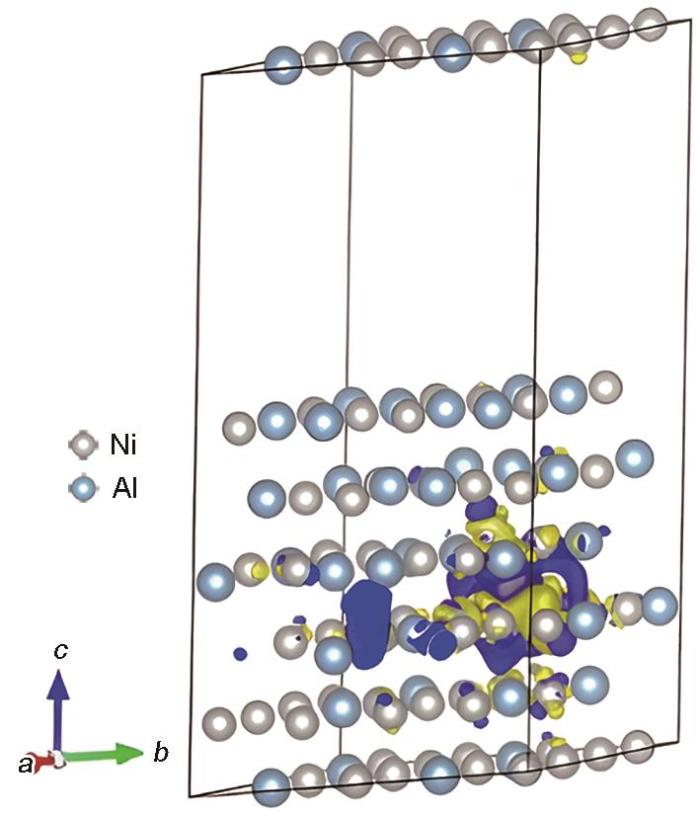
图9
含S元素Ni3Al/NiAl体系界面处二次差分电荷密度图
Fig.9
Differential charge density at the interface of Ni3Al/NiAl system within S (The yellow and blue regions represent the increase and decrease of charge density, respectively. The same in Fig.11)
图10
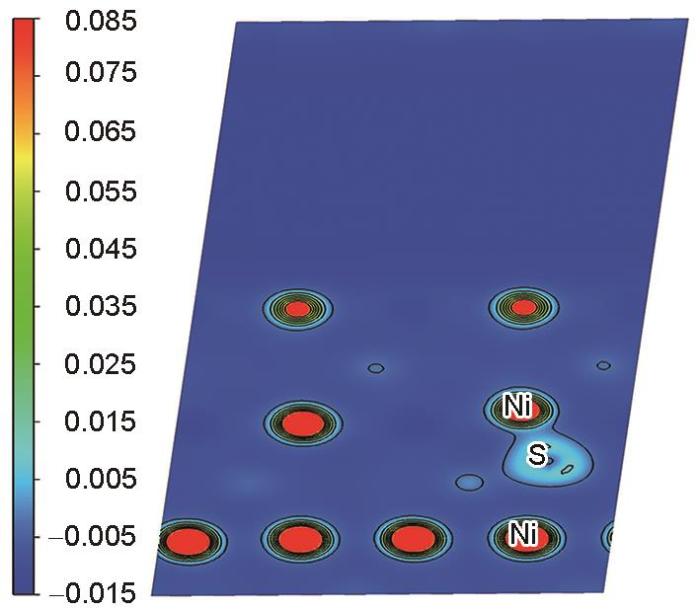
图10
Ni3Al/NiAl界面处S原子附近(010)面电荷密度分布
Fig.10
Distribution of (010) surface charge density near S atom at Ni3Al/NiAl interface (The blue area in the legend represents negative values, indicating the charge decrease, while the green to red area represents positive values, indicating the charge increase. The same in Figs.12, 17, and 18)
对于NiAl/Al2O3界面,二次差分电荷密度图见图11和12。因为有强电负性O原子的存在,使得S元素对于NiAl/Al2O3体系的影响与基体Ni3Al/NiAl涂层之间的影响有很大差别。从弛豫后的模型S原子的位置对比可知:S原子倾向位于Al2O3氧化层中,且S原子周围电荷密度减少。另外因O原子的强电负性,2者之间为蓝色区域(电荷密度减少),故S原子与其近邻的O原子存在一定的排斥作用。氧化层中与O结合的Al原子所需要的电荷也会增加,因此S原子的加入通过加速氧化层中Al元素电子的消耗对氧化层产生不利影响,进而影响NiAl/Al2O3界面结构的稳定性。另外,界面附近靠近NiAl一侧的Ni、Al原子周围电荷转移的区域减少,且S近邻的Ni原子附近电荷转移量较基体内层的Ni原子处少。因此S原子的加入还会破坏涂层一侧界面附近Ni、Al原子之间的电荷转移区域,使得涂层中Ni、Al原子之间连接的紧密程度降低。从图12的二维电荷密度图可知,S原子电荷密度略有降低,因为O原子的电负性更强,电荷主要聚集在其近邻的O原子周围。2者之间不发生电荷之间的转移,存在一定的排斥作用。对于NiAl一侧,清晰可见界面附近的Ni、Al原子间电荷转移量弱于内层(远离界面的) Ni、Al原子,界面附近Ni、Al之间的结合也会因此削弱。综上,S元素的存在不只对氧化层产生负面影响,也使得NiAl涂层原子之间的键合减弱,由此降低界面结合强度,最终对合金的性能产生影响。
图11
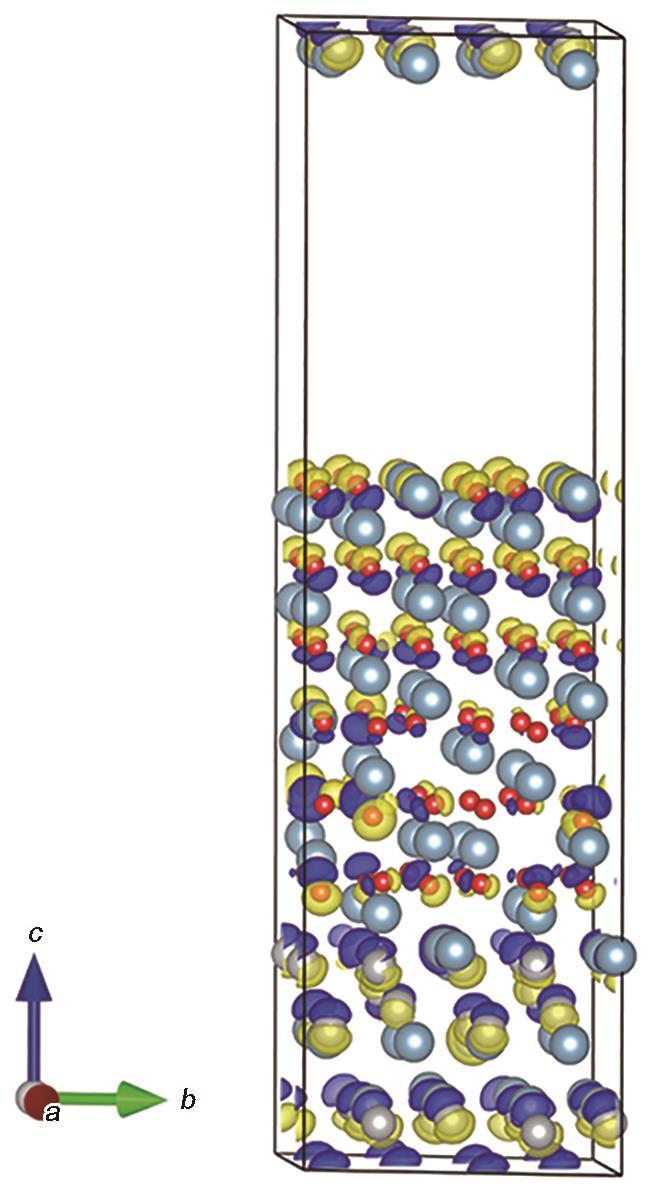
图11
含S元素NiAl/Al2O3体系界面处二次差分电荷密度图
Fig.11
Differential charge density at theinterface NiAl/Al2O3 system within S
图12
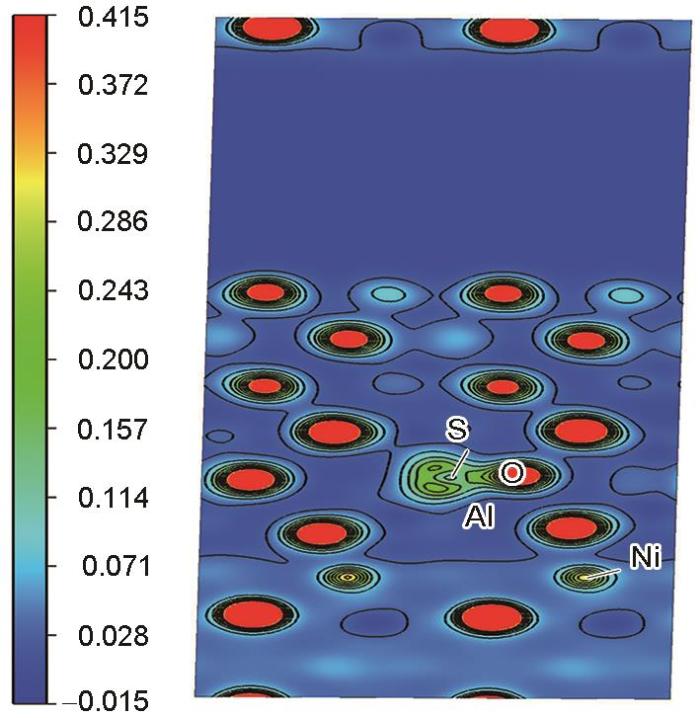
图12
NiAl/Al2O3界面处S原子附近(010)面电荷密度分布
Fig.12
Distribution of (010) surface charge density near S atom at NiAl/Al2O3 interface
2.4 电荷定量分析
通过呈现Ni3Al/NiAl界面体系中不同位置的Al、Ni原子电荷转移情况验证上述定性分析,模型中对应原子序号见图13,Bader电荷如图14所示。在Ni3Al/NiAl界面中,纯净界面位于基体Ni3Al中的21号Al原子在添加S元素前后电子转移能力变化最大,纯净界面原子电荷转移情况为-0.47e,含S原子的体系中Al原子的电荷转移情况为-0.440608e。Al原子转移出电荷的数量略有下降,下降值为0.027371e。同体系中其远离界面处的Al原子不同,界面附近位于Ni3Al基体中的12号Al原子、22号Al原子在含S原子的界面体系中,失去的电子数目基本保持不变。Ni原子同Al原子情况类似,在含S原子的体系中,界面附近Ni原子得到电子的数量减少,不同于较远离界面的Ni原子。其中界面两侧基体Ni3Al中的72号Ni原子和涂层NiAl中的76号Ni原子电子转移情况如下:纯净界面中,72号Ni原子电子转移情况为+0.171805e,76号Ni原子电子转移情况为+0.311233e。S原子存在的体系中,2者电荷转移情况分别为+0.163953e和+0.289884e,其接收电荷数量分别减少了0.007852e和0.021349e。S原子使体系界面附近S近邻的基体中Al原子失电子能力减弱,因此S元素对基体Ni3Al和NiAl涂层的影响体现在使界面S原子近邻的Ni、Al原子之间的电荷转移量降低(Al原子失去的电子减少,Ni得到的电子减少)。为了更全面地说明电荷转移的整体情况,还对整个界面体系中S近邻(做弛豫的几层原子)进行了得失电子的总体统计(如表3所示,正值代表得到电子,负值代表失去电子)。界面附近58个Ni原子在无S和含S界面体系中得到的电子数分别为9.974558e和10.112086e,界面附近36个Al原子无S和含S界面体系中失去的电子数分别为8.673444e和8.661022e。含S体系中界面附近弛豫的Ni原子总计转移电荷(得到的电子)增加了0.137528e,Al原子总计电荷转移变化(失去的电子)较Ni的少,总失去的电子减少了0.012422e。对于S原子,加入到体系中,其本身失去电子0.986231e,由此可以解释整体Ni原子得到电子数目的增加:S失去的电子转移到了Ni之中,参与Ni—S之间的成键,Ni原子电子转移量的增大,导致原子杂化程度增大,界面稳定性逐渐降低[39],这也会进一步导致Ni—Al之间的结合紧密程度下降。
图13

图13
Ni3Al/NiAl体系中部分原子序号
Fig.13
Partial atomic numbers in the Ni3Al/NiAl system
图14
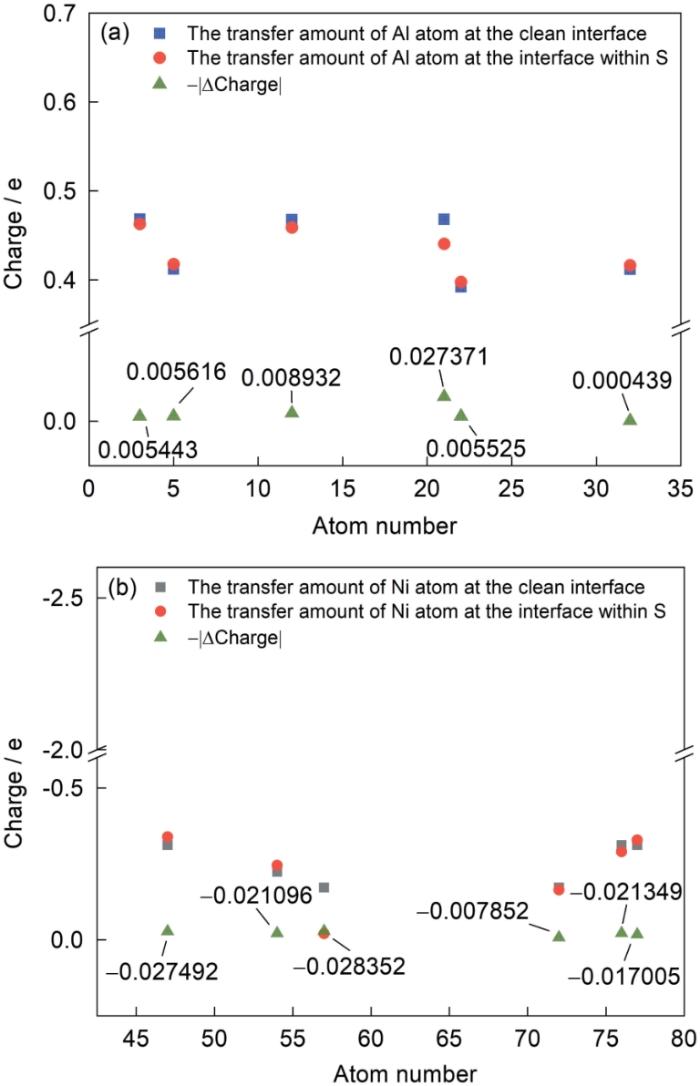
图14
Ni3Al/NiAl界面近邻原子Bader电荷
Fig.14
Bader charge transfer between adjacent atoms at Ni3Al/NiAl interface (Charge—Bader charge values of the designated atom, atom number—the number of designated atoms in corresponding system)
(a) Al atom (b) Ni atom
表3 Ni3Al/NiAl界面体系Bader电荷转移的统计情况
Table 3
| Atom | Bader value | |
|---|---|---|
| Pure interface system | Interface system within S | |
| Ni | 9.974558e | 10.112086e |
| Al | -8.673444e | -8.661022e |
| S | -0.986231e | |
对于NiAl/Al2O3界面体系,通过呈现无S和含S元素体系中特定原子的电子转移量以印证差分电荷密度部分的分析。为了获得更为全面的电子分析,也对S原子近邻的几层弛豫原子层进行了得失电子总量的统计。模型中对应原子序号见图15,Bader电荷如图16所示。图16a中界面附近氧化层中的16、18、28、30号Al原子在含S原子的界面体系中呈现出失电子减少的趋势,其中S近邻的28、30号原子变化趋势较其他原子大,失去电子减少的数值分别为0.237910e和0.190981e。对于图16b在涂层中的Ni原子结果显示;界面附近的84、91号Ni原子在含S原子界面体系中呈现得电子增加的趋势,其他原子在含S原子的体系中得到的电子均减少。由图16c中对比无S和含S的界面体系氧化层中O原子电子转移情况:发现与S近邻的112号O原子电子转移变化最大,其得到的电子减少了0.057951e。由此可见,S与O强烈的排斥作用,使得氧化层中O原子电负性略有减弱。对体系进行电子转移数量统计(表4),结果显示S原子存在的界面体系中,界面两侧近邻(弛豫的原子层)中,Al、Ni原子失去/得到的电子都呈现减少的趋势,数值分别为0.470627e和0.004057e,S原子附近的O原子得到电子的数量增加了1.161669e。S原子失去电子0.512175e,结合二次差分电荷密度的可视化结果,可知由于Al2O3层的存在,电荷转移主要发生在Al—O以及Ni—Al之间,且加上S原子与O原子之间的排斥作用,S失去的电子聚集在O原子周围,因此S元素的存在会加大氧化层内Al原子中电子的消耗,同时由于S原子影响近邻Ni原子中电子的转移,进一步使得Al原子中电子转移数量下降,最终降低NiAl涂层中Ni、Al原子间连接紧密程度,使得该界面体系中键连接紧密性下降。
图15
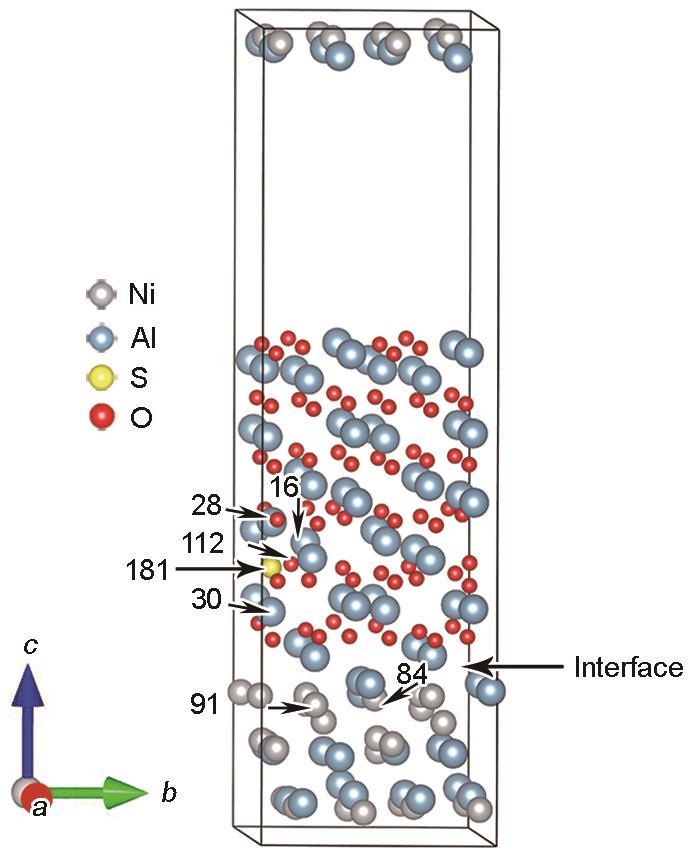
图15
NiAl/Al2O3体系中部分原子序号
Fig.15
Partial atomic numbers in the NiAl/Al2O3 system
图16
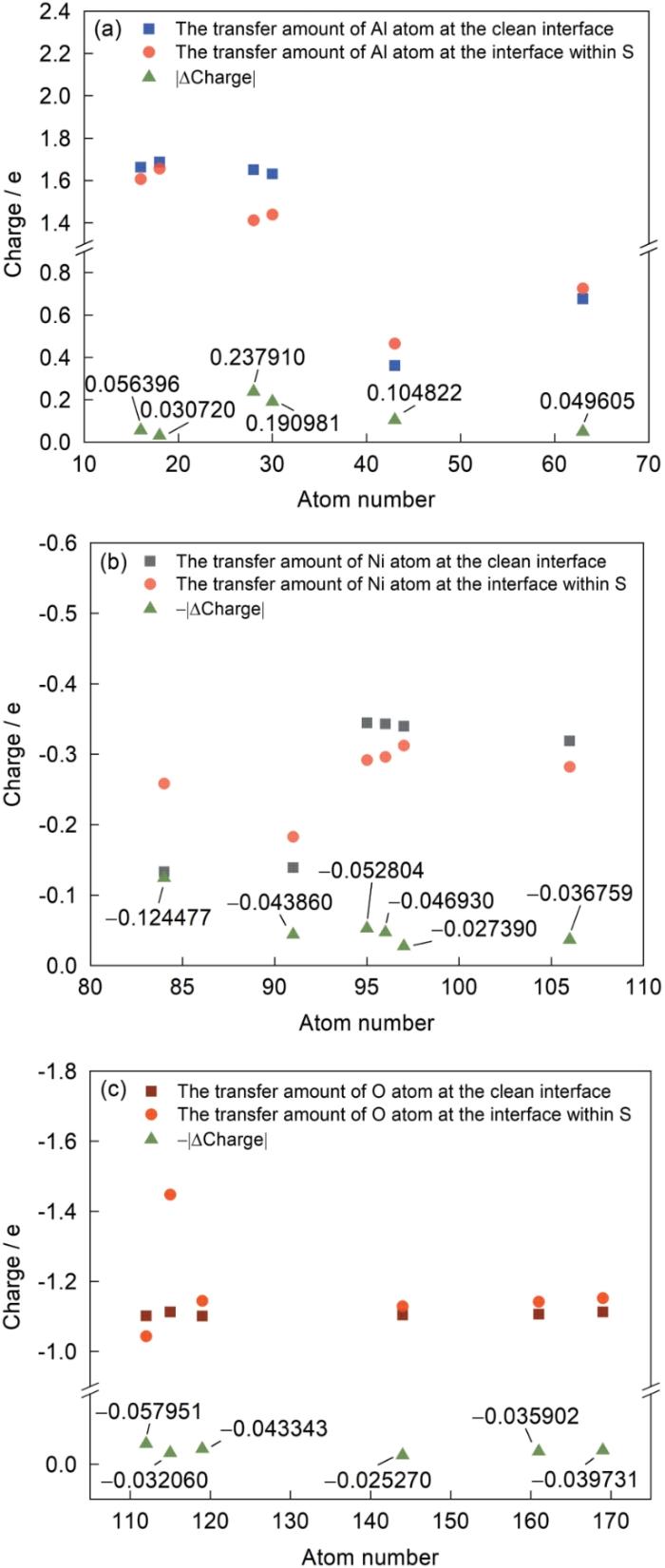
图16
NiAl/Al2O3界面近邻原子Bader电荷转移
Fig.16
Bader charge transfer between adjacent atoms at NiAl/Al2O3 interface
(a) Al atom (b) Ni atom (c) O atom
表4 NiAl/Al2O3界面体系Bader电荷转移的统计情况
Table 4
| Atom | Pure interface system | Interface system within S |
|---|---|---|
| S | -0.512175e | |
| Ni | 7.239174e | 7.235117e |
| Al | -51.712821e | -52.242194e |
| O | 57.058943e | 58.220612e |
2.5 ELF分布图
Ni3Al/NiAl纯净界面的ELF分布见图17a,含S元素Ni3Al/NiAl界面的ELF分布见图17b,为观察与S近邻原子的二维ELF分布图,2者与二次差分电荷密度所选的平面位置一致,即选取(010)面进行投影。可以看出:界面两侧各自的体系中Ni原子和Al原子之间呈现出共价键的特征,在涂层NiAl一侧还存在离子键的特征。然而同体系中含S原子后,界面附近S原子近邻的Ni原子周围蓝色区域有所减少,S原子与近邻的Ni原子存在电荷的聚集区,含S体系中发生的原子间电荷的变化导致了体系中原有的离子键、共价键被削弱,即界面附近局部连接键合变得不紧密,从键合角度解释了界面黏附功降低的微观机制。图中S原子周围主要是红色的区域,而S原子附近的Ni原子呈现蓝色区域,可见电子从Ni原子离域,转移到S原子周围,2者之间形成离子键;因此会降低涂层中Ni、Al原子间共价键键合的紧密性,在基体中也存在共价键被削弱的现象,而这会导致基体Ni3Al致脆以及影响NiAl的韧性。此外还可以印证Bader电荷分析中:S元素的存在使体系界面附近S近邻的基体中Al、Ni原子失/得电子的能力减弱的分析结果。
图17
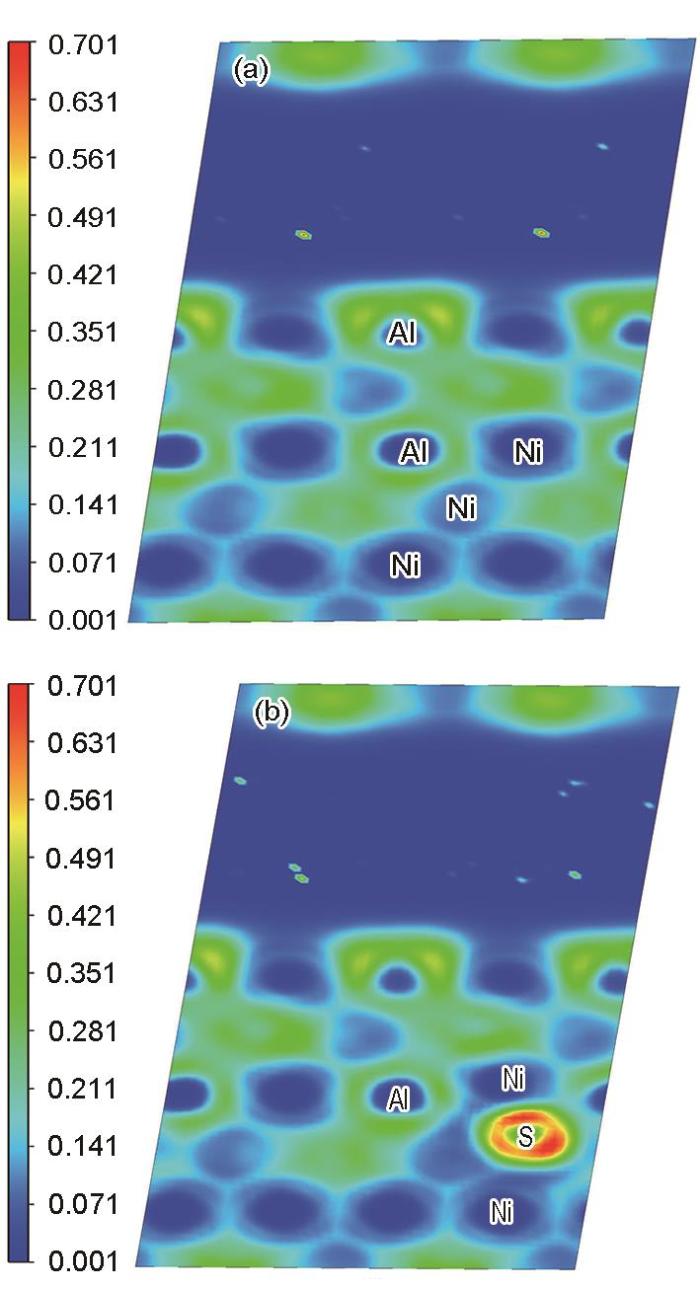
图17
Ni3Al/NiAl体系(010)截面的电子局域化函数(ELF)投影图
Fig.17
Electron localization function (ELF) projection in (010) plane of Ni3Al/NiAl system
(a) clean (b) within S element
涂层NiAl/氧化层Al2O3界面绘制的(010)处ELF分布如图18所示,进一步分析界面体系中Ni—Al、Al—O之间在无S和含S元素Ni3Al/NiAl界面体系中的电子局域化函数分布及成键特性。图中ELF值较大的主要在O原子和S原子周围,因O原子的电负性较强,其局域性也强于S原子,电子有聚集在其周围的趋势,由此也对应了电荷密度的分析结果。S、O原子之间的区域说明两原子间存在的排斥作用。在氧化层中,Al原子周围ELF值接近为0,而与其相邻的O原子ELF值较高(见红色区域),Al原子中的电子离域化移向了O原子,2者间形成较强的离子键。然而在含S的体系中,S原子近邻的O原子周围电子局域性增加,与其形成离子键的Al原子离域性有所增加,表明Al、O原子间形成键合所需要的电子增加,会消耗掉比纯净体系更多Al原子的电子,加大氧化层的消耗。对于NiAl涂层,因为S原子的存在,界面附近Al原子的离域趋势有所降低,原因可能在于氧化层中的氧化不断消耗Al元素,同时伴随着在界面体系处发生的元素扩散导致β-NiAl涂层向γ'相的转变[40],可用来提供Al电子的Al元素含量降低。Audigié等[41]通过实验研究得出,在界面附近涂层和互扩散区之间的扩散通量不平衡时,就会在界面附近产生空位并形成Kirkendall孔洞,而S元素在界面体系的偏聚有助于孔洞的形核和长大。
图18
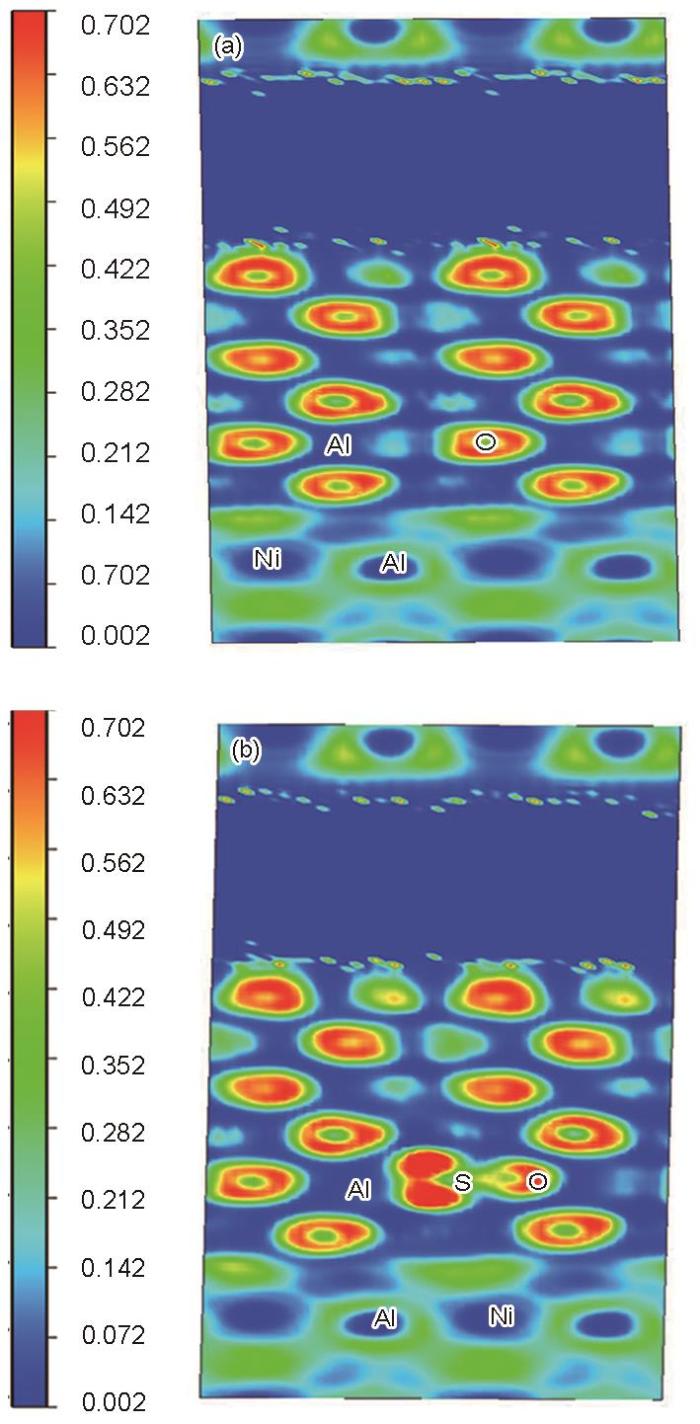
图18
NiAl/Al2O3体系(010)截面的ELF投影图
Fig.18
ELF projection in (010) plane of NiAl/Al2O3 system
(a) clean (b) within S element
2.6 态密度图
为进一步了解S原子作用下界面原子间轨道相互作用情况,运用数据处理得到了2种界面体系(Ni3Al/NiAl、NiAl/A2O3)的TDOS和LPDOS,见图19~22,图中每个横坐标上的零能量点代表系统的Fermi能级,Ni′、Al′、O′对应情况为含S元素的界面体系。将纯净/含S界面体系的TDOS作为对照,以探究S元素的存在对界面体系的影响,此外也分别呈现了界面附近S原子近邻的
图19
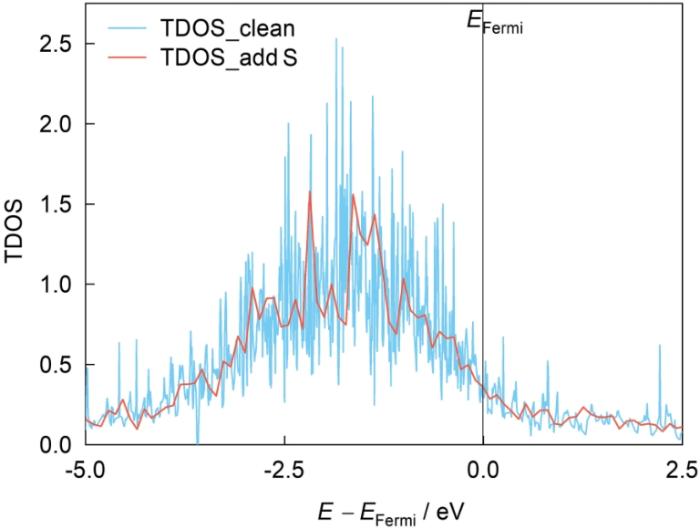
图19
Ni3Al/NiAl界面体系的总态密度(TDOS)
Fig.19
Total density of states (TDOS) of Ni3Al/NiAl interface system (EFermi—Fermi energy, E—energy)
图20
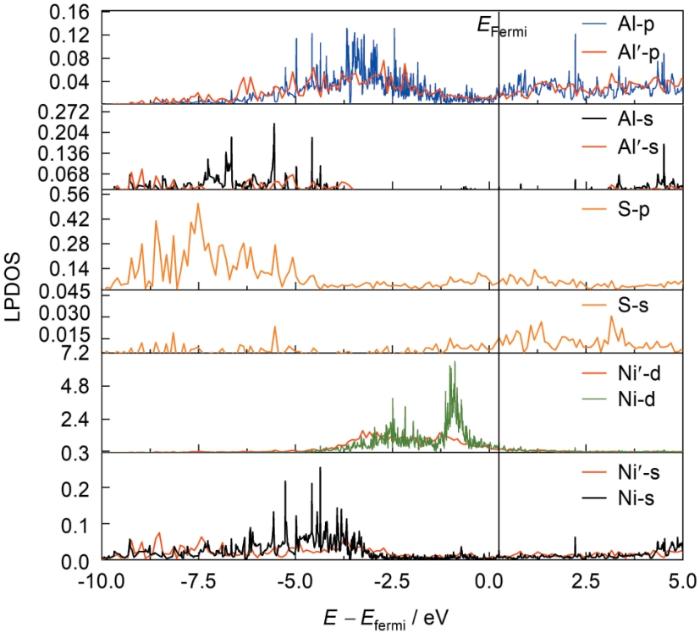
图20
Ni3Al/NiAl界面体系S原子及其近邻Al、Ni原子的局域分波态密度(LPDOS)图
Fig.20
Local partial density of states (LPDOS) of S atom and its neighboring Al, and Ni atom in Ni3Al/NiAl interface system (Ni', Al', and O' correspond to interface systems containing S elements)
图21
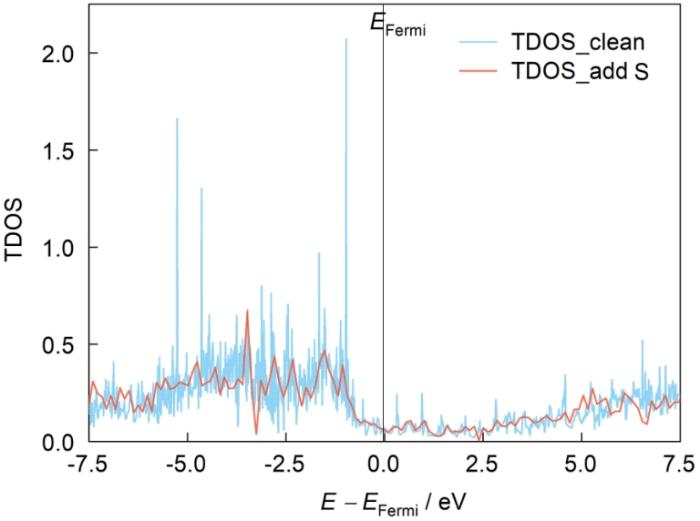
图22

图22
NiAl/Al2O3界面体系S原子及其近邻
Fig.22
LPDOS of S atom and its neighboring
图21为NiAl/Al2O3界面模型体系的总态密度分布。可见在-7.5~0 eV之间态密度的峰值下降程度很大,在0~7.5 eV之间态密度延展分布,峰值展宽,说明在涂层NiAl/氧化层Al2O3界面附近由于强电负性S原子的存在,S与O原子之间存在电荷聚集,因此态密度峰尖锐程度有所降低。对于-7.5~0 eV区间,主要是Ni的d电子离域性增加,导致峰值下降,说明S元素的存在间接影响涂层中Ni电子的转移,其与S相互作用分散了Ni与Al原子之间电子转移的数量,进一步降低涂层中Ni、Al之间键结合的紧密程度,这与电子局域密度的分析结果一致。NiAl/Al2O3体系中无S与含S体系
3 结论
(1) S对镍基高温合金的影响机理:从能量角度来看,S元素在合金中会使得界面黏附功降低约0.96%,使界面体系变得不稳定,且S元素有较强的界面偏聚特性。S元素的存在导致基体Ni3Al原子结合键削弱,进一步会影响涂层NiAl的韧性,此外S原子与近邻NiAl涂层中的Ni原子之间形成离子键,导致合金中Ni、Al原子之间局部金属键的连接紧密程度减弱,同理导致涂层中Ni、Al共价键的削弱,最终影响合金的性能。态密度的结果进一步解释了共价键被削弱的原因:总态密度显示S原子的存在使得近邻的Ni原子d电子的局域性减弱;局域分波态密度表明Ni原子的s、p、d轨道的峰值均有所下降(其中Ni-d轨道的下降趋势更大),以上进一步导致了参与Ni—Al之间成键电子的数量减少,使得基体中Ni、Al原子之间共价键结合减弱。
(2) S对镍基高温合金热障涂层的影响机理:S元素在界面处的偏聚也会降低基体/氧化膜的界面结合强度。S元素在体系中使得界面黏附功降低了8.98%,为维持自身稳定性也具有偏聚到涂层一侧的特性。经电子结构分析阐明了S元素影响界面稳定性的微观机制:界面附近S原子与近邻O原子的电荷密度均呈现增加,且2者之间存在一定排斥作用。一方面S元素的加入会加速氧化膜中Al元素中电子的消耗,进而影响NiAl/Al2O3的界面结构的稳定性,降低界面黏附功;另一方面S原子的加入还会破坏涂层一侧界面附近Ni、Al原子之间的电荷转移区域,从总态密度和局域分波态密度的角度分析得知:体系的总态密度峰尖锐程度有所降低,且Ni原子的d电子离域性增加导致峰值明显下降,说明S元素的存在间接影响涂层中Ni电子的转移,进一步降低涂层中Ni、Al之间键结合的紧密程度,最终削弱涂层自身的性能;分波态密度中可见S元素存在引起的显著变化:Ni'-d向Fermi能级移动(增加系统的能量),从而降低了体系中界面的稳定性。
参考文献
Microstructure evolution behavior of powder superalloy FGH4720Li at near service temperature
[J].
GH4720Li is used for turbine disks in a large number of civil and military propulsion systems because of its excellent mechanical properties and corrosion resistance. GH4720Li turbine disk is mainly manufactured through cast and wrought conventionally, but the addition of a high mass fraction of Ti, Al, and Mo can cause severe element segregation and more difficult microstructure control, which can become more severe as the turbine disk gets larger. Owing to this difficulty, the turbine disk quality cannot be guaranteed if the GH4720Li turbine disk is still in cast and wrought form, and the manufacturing process will be more complex, resulting in increased costs. However, the powder metallurgy method can efficiently eliminate element segregation and produce a more uniform microstructure than the cast and wrought methods. GH4720Li alloys manufactured using the powder metallurgy method are called FGH4720Li alloys. As there has been limited research on FGH4720Li and no report on the microstructure evolution during long-term ageing for FGH4720Li to date, it is necessary to study the microstructure evolution behavior during long-term ageing for FGH4720Li to obtain improved microstructure stability. In this study, field emission scanning electron microscopy and extractive phase analysis were used to investigate the microstructure evolution of FGH4720Li in the temperature range of 600-730oC up to 3000 h. The results showed that primary gamma prime was the most stable; whereas, secondary and tertiary gamma prime microstructure evolutions were comparatively complex. At 600oC, there was no microstructure change. At 650oC, only the tertiary gamma prime grew, but there was no microstructure change for the other gamma primes up to 3000 h. When the ageing temperature increased to 730oC, the tertiary gamma prime grew faster before coarsening rapidly. After 200 h, the secondary gamma prime coarsened noticeably, but the big secondary gamma prime coarsened by Ostwald ripening first, absorbing a large amount of tertiary gamma prime and splitting up between 300 and 500 h, before ageing with further processing. Big secondary gamma prime mainly coarsens by amalgamation; whereas, small secondary gamma prime always coarsens by amalgamation during ageing. The divergence between these two types of secondary gamma prime is related to the distribution characteristics of the tertiary gamma prime.
粉末高温合金FGH4720Li在近服役温度下的组织演变规律
[J].
The feasibility and process control of uniform equiaxed grains by hot deformation in GH4720Li alloy with millimeter-level coarse grains
[J].For nickel-based GH4720Li superalloys, fine-grained structures can be obtained via hot deformation in a two-phase area. However, obvious recrystallized grains' coarsening occurs because of the lack of prime γ' pinning grain boundary when hot deformation occurs in a single-phase area. Much attention is directed to the hot deformation of GH4720Li alloys with fine grains. Moreover, several studies have reported the hot deformation behavior of coarse-grained GH4720Li alloys, with maximum grain sizes of several hundred microns. However, only a few studies report about the hot deformation of GH4720Li alloy with millimeter-level coarse grains. Coarse grains recrystallize incompletely and can reduce the hot deformation plasticity of GH4720Li alloy. Thus, to clarify the coordination feasibility between recrystallization and the hot deformation plasticity of GH4720Li alloy with millimeter-level coarse grains, the hot deformation behavior of GH4720Li alloy with millimeter-level coarse grains was investigated under different deformation parameters (deformation temperature of 1130, 1160, and 1190°C; strain rates of 0.001, 0.01, 0.1, and 1 s-1; and engineering strain of 50%) and compared with the hot deformation behavior of fine-grained GH4720Li alloy. The results show that the GH4720Li sample with millimeter-level coarse grains is more sensitive to the deformation temperature, but the fine-grained GH4720Li alloy is more sensitive to strain rate. The completely recrystallized structure of the GH4720Li sample with millimeter-level coarse grains can be obtained in the range of 1160-1190°C and 0.001-0.01 s-1. However, a meager strain rate can cause an undesirable and obvious grain growth. After comprehensively combining the recrystallization control range and the hot deformation plasticity of millimeter-level coarse-grained GH4720Li alloy, it was found that the millimeter-level coarse-grained GH4720Li alloy should be thermally deformed at a moderate deformation temperature and a reduced strain rate of 1160oC and 0.01 s-1, respectively, to obtain the uniform equiaxed grains structure without cracking, and better deformation.
GH4720Li合金毫米级粗大晶粒热变形获得均匀等轴晶粒的可行性及工艺控制
[J].
Effect of micron-sized particles on the crack growth behavior of a Ni-based powder metallurgy superalloy
[J].
Effect of overheating events on microstructure and low-cycle fatigue properties of a nickel-based single-crystal superalloy
[J].
Thermal barrier coatings for gas-turbine engine applications
[J].Hundreds of different types of coatings are used to protect a variety of structural engineering materials from corrosion, wear, and erosion, and to provide lubrication and thermal insulation. Of all these, thermal barrier coatings (TBCs) have the most complex structure and must operate in the most demanding high-temperature environment of aircraft and industrial gas-turbine engines. TBCs, which comprise metal and ceramic multilayers, insulate turbine and combustor engine components from the hot gas stream, and improve the durability and energy efficiency of these engines. Improvements in TBCs will require a better understanding of the complex changes in their structure and properties that occur under operating conditions that lead to their failure. The structure, properties, and failure mechanisms of TBCs are herein reviewed, together with a discussion of current limitations and future opportunities.
Research progress in materials and preparation techniques of thermal barrier coatings
[J].
热障涂层材料及其制备技术的研究进展
[J].
Recent development in materials design of thermal barrier coatings for gas turbine
[J].
航空发动机热障涂层材料体系的研究
[J].介绍了热障涂层的结构、陶瓷表层材料和中间粘结层的材料发展趋势。热障涂层的结构已由经典的双层结构向成分、结构连续变化的梯度结构发展,涂层的寿命有明显的提高。制备方法中电子束物理气相沉积技术具有明显优势。热障涂层表层材料的最佳成分是(质量百分数6%~8%)Y2O3部分稳定的ZrO2陶瓷材料。粘结层体系发展的一个重要趋势是能同时保证力学性能和抗氧化性能的低Al含量NiCoCrAlY。
Studies of the bond-coat oxidation and phase structure of TBCs
[J].
Effect of ppm level sulfur addition on isothermal oxidation behavior of a nickel-base single crystal superalloy
[J].
ppm级S对第二代抗热腐蚀镍基单晶高温合金恒温氧化行为的影响
[J].
The segregation of sulfur and phosphorvs in nickel-base alloy 718
[J].
Chemical and morphological evolution of a (NiPt)Al bond coat
[J].
Characterization of chemical and microstructural evolutions of a NiPtAl bondcoat during high temperature oxidation
[J].
Sulfur localization in NiPtAl/superalloy systems after high temperature isothermal oxidation
[J].
Effects of additives and impurity on the adhesive behavior of the NiAl(110)/Al2O3 (0001) interface: An ab initio study
[J].
Molecular dynamics simulation of the γ′ phase deformation behaviour in nickel-based superalloys
[J].
Sulfur embrittlement on γ/γ' interface of Ni-base single crystal superalloys
[J].
Characterisation of oxide scale adherence after the high temperature oxidation of nickel-based superalloys
[J].
Spallation and transient oxide growth on PWA 1484 superalloy
[J].
Bayesian optimization for calibrating and selecting hybrid-density functional models
[J].
From ultrasoft pseudopotentials to the projector augmented-wave method
[J].
Generalized gradient approximation made simple
[J].
Atomic size effect of alloying elements on the formation, evolution and strengthening of γ′-Ni3Al precipitates in Ni-based superalloys
[J].
Effects of segregating elements on the adhesive strength and structure of the α-Al2O3/β-NiAl interface
[J].
Extracting convergent surface energies from slab calculations
[J].
First-principles studies of effects of interstitial boron and carbon on the structural, elastic, and electronic properties of Ni solution and Ni3Al intermetallics
[J].
First-principles study of Ni/Ni3Al interface doped with Re, Ta and W
[J].
Adhesion in NiAl-Cr from first principles
[J].
Surface segregation of Si and its effect on oxygen adsorption on a γ-TiAl(111) surface from first principles
[J].
First-principles study of the strengthening mechanism of Fe-Cr-Al alloy interface
[D].
Fe-Cr-Al合金界面强化机理的第一性原理研究
[D].
First-principles study of the effect of S impurity on the adhesion of Fe/Al2O3 interface
[J].
杂质S对Fe/Al2O3界面结合影响的第一性原理研究
[J].
Effects of Zr-Re/W Co-segregation behavior on the thermodynamic stability and fracture strength of γ-Ni/γ'-Ni3Al interface
[J].
Ab initio calculations of the 3C-SiC(111)/Ti polar interfaces
[J].
An atomistic investigation into the nature of fracture of Ni/Al2O3 interface with yttrium dopant under tension
[J].
Formation of aluminides on Ni-based superalloy 690 substrate, their characterization and first-principle Ni(111)/NiAl(110) interface simulations
[J].
First-principles determination of the effects of boron and sulfur on the ideal cleavage fracture in Ni3Al
[J].
Current research status of interface of ceramic-metal laminated composite material for armor protection
[J].A composite material with a laminated structure can be fabricated through layer-by-layer stacking of ceramic and metal in a certain order. It has characteristics of high strength, high hardness, low density of ceramics, and strong ductility of metals; thus, it can be used for bulletproof armor materials. During bullet antipenetration, the ceramic panel is responsible for decelerating and breaking the projectile, and the metal backplate absorbs the kinetic energy of the bullet through plastic deformation, thereby forming a complete bulletproof armor system. However, there are some problems associated with laminated materials, such as the significant difference between the properties of ceramic and metal, weak interface bonding strength, and easy occurrence of tip cracks due to the increase in the internal stress of the impacted material. The ceramic-metal interface easily leads to a sudden change in material properties, and crack propagation and migration affect the properties. After being impacted, cracks first appear in the interlayer, where the interface bonding strength is still unideal, easily leading to a drop between the ceramic panel and metal backplate. In this study, the preparation and observation of interface structure, phase-field simulation of interface fracture, finite element simulation of interface impact resistance, meshless smoothed-particle hydrodynamic method for high-velocity impact and large deformation, and first-principles calculations of interface strength were reviewed. Finally, some suggestions are presented for future development: (1) Strengthening the research of ceramic toughening to enhance the matching degree between the ceramic panel and metal backplate, reducing the sudden change of ceramic to metal performance, and making the performance of ceramic-metal laminated materials more uniform is crucial. In addition, studying metal strengthening is necessary. On the premise of not damaging metal ductility, nano-phases can be added to prepare metal matrix composites for metal strengthening; (2) More multiscale research methods, such as phase-field method, finite element analysis, and first-principles calculations are needed, especially focusing on how to organically and effectively combine these methods. The complementary coupling of multiscale experimental research and computational simulation methods is a powerful tool for the interface design of ceramic-metal laminated materials in the future.
装甲防护陶瓷-金属叠层复合材料界面研究进展
[J].将陶瓷与金属以一定顺序逐层叠加,可制成叠层结构的复合材料,兼具陶瓷高强度、高硬度、低密度及金属强延展性的特点,从而应用于防弹装甲材料。但叠层材料存在界面结合弱、受冲击时裂纹易在界面处产生,且裂纹尖端应力集中导致界面处材料易脱黏等问题。本文针对陶瓷-金属叠层复合材料的界面结构及结合强度的问题,从界面结构的制备和观察、界面断裂的相场模拟、界面抗冲击性的有限元模拟和界面强度的第一原理计算等方面进行了综述,并对未来发展方向提出建议。
The effect of sulfur segregation on the adherence of the thermally-grown oxide on NiAl—I: Sulfur segregation on the metallic surface of NiAl(001) single-crystals and at NiAl(001)/Al2O3 interfaces
[J].
Stability of phase boundary between l12-Ni3Al phases: A phase field study
[J].
Service damage mechanism and interface cracking behavior of Ni-based superalloy turbine blades with aluminized coating
[J].
Observation and modeling of α-NiPtAl and kirkendall void formations during interdiffusion of a Pt coating with a γ-(Ni-13Al) alloy at high temperature
[J].
Site preference and brittle-ductile transition mechanism of B2-NiAl with ternary elements additions form first-principles calculations
[J].
Structural stability and mech-anical property of Ni(111)-graphene-Ni(111) layered composite: A first-principles study
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}