


Al-Li合金具有高弹性模量、低密度、高比强度、高比刚度以及良好的抗疲劳扩展等优势,广泛用于航空、航天领域[1~3]。析出强化作为Al-Li合金强度、韧性力学性能调控的重要手段,广泛用于新型Al-Li合金的开发、设计。其中主要围绕合金成分设计、变形预处理以及时效热处理等冶金工艺用于析出相的种类、含量、分布的调控。与第一、二代Al-Li合金相比,第三、四代Al-Li合金的元素种类以及各组元含量都有大的改变。主要呈现出Li含量降低、Cu含量提高,同时添加其他微合金元素,一般包含Zn、Cr、Mn、Zr、Mg、Ag、Ti、Sc、B以及稀土元素La、Ce和Y等[4~14],用于特定需求的Al-Li合金开发。不同微合金元素的添加对纳米级沉淀相的析出起到不同的影响,例如Ag、Mg原子在T1相(Al2CuLi)与基体界面处偏析可以降低T1相形核势垒[12];Cu原子与空位形成的团簇结构在时效过程中逐渐转变为GP区、亚稳θ'(Al2Cu)以及稳定的θ (Al2Cu)相[11]。Mn、Sc和Zr原子倾向在θ'相与基体共格、半共格界面处偏析。这些合金元素改善了θ'相在高温下的稳定性;对于θ'相的形核、生长和粗化也产生重要的影响[13];Sc原子与Al原子形成与基体共格的L12-Al3Sc相,先析出的Al3Sc相作为δ'相(Al3Li)的异质形核位点,可以形成核(Al3Sc)-壳(Al3Li)结构[14]。事实上,纳米析出相的合理调控已成为打破传统金属材料强度、韧性不可兼得的有力途径。
在Al-Li合金中除T1相外,δ'/θ'/δ'复合沉淀相作为一种重要的强化相,已有广泛的理论与实验报道[15~19]。与传统Al-Cu合金中的θ'相不同的是,δ'/θ'/δ'复合沉淀相中的θ'相的厚度仅有几个纳米,表现出良好的抗粗化性能。考虑到θ'相在Al基体中的不可剪切性,相比易与Al基体产生共面滑移的δ'相,纳米级θ'相表现出更高的强化效果。结构上,δ'/θ'/δ'复合沉淀相是一种由透镜状δ'相在θ'相两端异质形核构成的一种三明治复合结构。研究[15~18]发现,该复合相两端δ'相伴随内部θ'相的生长存在同相(in-phase)和反相(anti-phase) 2种位相关系,表现为偶数Cu层时为反相结构;奇数Cu层则为同相结构。GP-I作为θ'相的一种“前驱”相,类似的结构特性也存在于δ'/GP-I/δ'复合结构中[15]。第一性原理计算结果[17]表明,δ'/θ'界面结构的界面能相比于θ'/Al和δ'/Al界面能更低,使得δ'相在θ'相上的异质形核成为可能。然而不论是利用透射电子显微镜(TEM)还是先进的高角环形暗场扫描透射电子显微镜像(HAADF-STEM)技术,对于复合沉淀相δ'/θ'/δ',其θ'/δ'界面终端的原子排列、结合方式、两端δ'存在的同相和反相的相位结构特征以及可能的转变途径都是难以获得的。
基于此,本工作研究了δ'/θ'/δ'复合沉淀相中两侧δ'位相关系与θ'/δ'界面结构的联系,以及随着内层θ'相生长,同相和反相结构相互转变的方式。首先,从能量角度出发,确定出与实验观察结果一致且更稳定的同相结构;随后,构建可能存在的反相结构,通过滑移势能面和激活能计算,得出由同相转变为反相的最优途径;通过计算不同θ'/δ'界面结构的界面能,得出两侧δ'位相关系与δ'相在θ'相上异质形核的关系;分析界面黏合处的键能-位移(binding energy-displacement)关系,得到不同θ'/δ'界面结合方式的理想解理应力(ideal cleavage stress)[20]。最后,基于Hamilton矩阵的晶体轨道布居理论(COHP)[21],分析了界面结构中连接界面的原子对的成键机理。讨论了不同分子轨道的成、反键作用,以及对整体界面结构成键强度的贡献。本工作对于理解δ'/θ'/δ'复合沉淀相的形核、生长具有启发意义,为设计高强Al-Li合金提供了新思路。
1 计算模型与方法
2 计算结果与讨论
2.1 同相和反相结构
图1
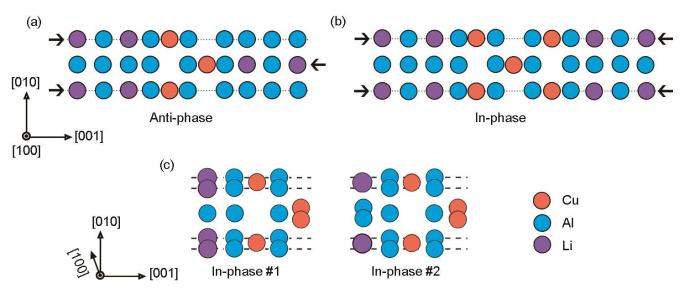
图1
δ'/θ'/δ'复合沉淀相中反相和同相结构示意图以及2种同相的界面结构
Fig.1
Schematics of anti-phase (a) and in-phase (b) in the δ'/θ'/δ' composite precipitate, and two kinds of in-phase interfacial structures (c) (The arrows are used to assist in showing the relationship for the opposite δ' phases)
为了选取稳定的同相界面结构,构建了包含不同Cu层数的复合沉淀相的结构模型,计算ΔH,计算公式如下[18]:
式中,E(Al n Cu b Li c )是复合沉淀相0 K下的总能;E(Al)、E(Cu)和E(Li)分别为组成元素Al、Cu和Li在平衡晶体结构下单个原子的能量;n、b和c分别为组成复合沉淀相Al、Cu和Li的原子个数。δ'/θ'/δ'复合沉淀相包含不同层Cu时,采用同相#1和同相#2界面结构计算所得的ΔH如图2所示。可以看出,不论是奇数还是偶数层Cu原子,#2都是能量上更稳定的同相界面结构。
图2
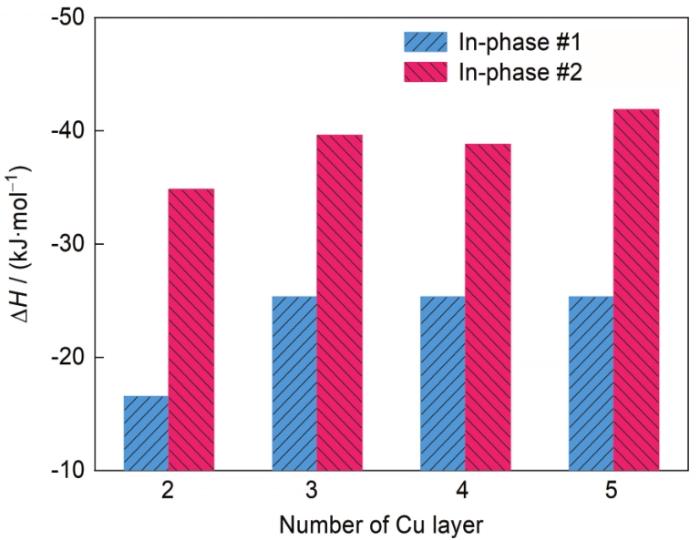
图2
δ'/θ'/δ'复合沉淀相包含不同层Cu时采用同相#1和同相#2界面结构计算所得的形成焓(ΔH)
Fig.2
Calculated formation enthalpies (ΔH) of the composite precipitate δ'/θ'/δ' containing different Cu layers with in-phase #1 and in-phase #2 two interfacial structures
图3
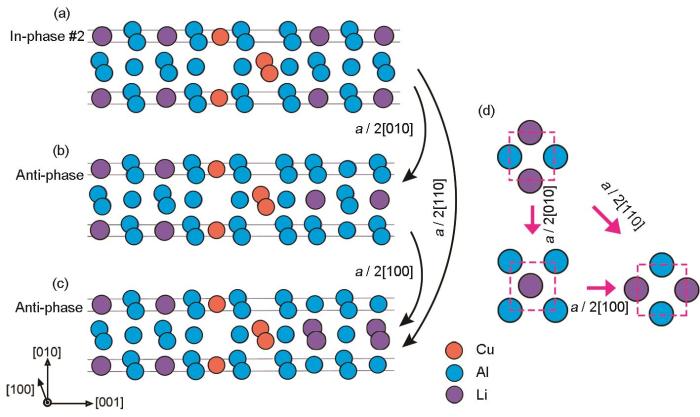
图3
δ'/θ'/δ'复合沉淀相由同相#2转变为反相a /
Fig.3
Schematics showing the transformation of the composite precipitate δ'/θ'/δ' from in-phase #2 (a) to anti-phase a /
为了给出由同相#2结构转变为这2种反相结构的转变路径,计算了不同转变方式对应的滑移势能面以及沿不同滑移方向转变需要的激活能(Eactivation),只考虑相界面的刚性运动,分别如图4a和b所示。可以发现,相比反相a /
图4
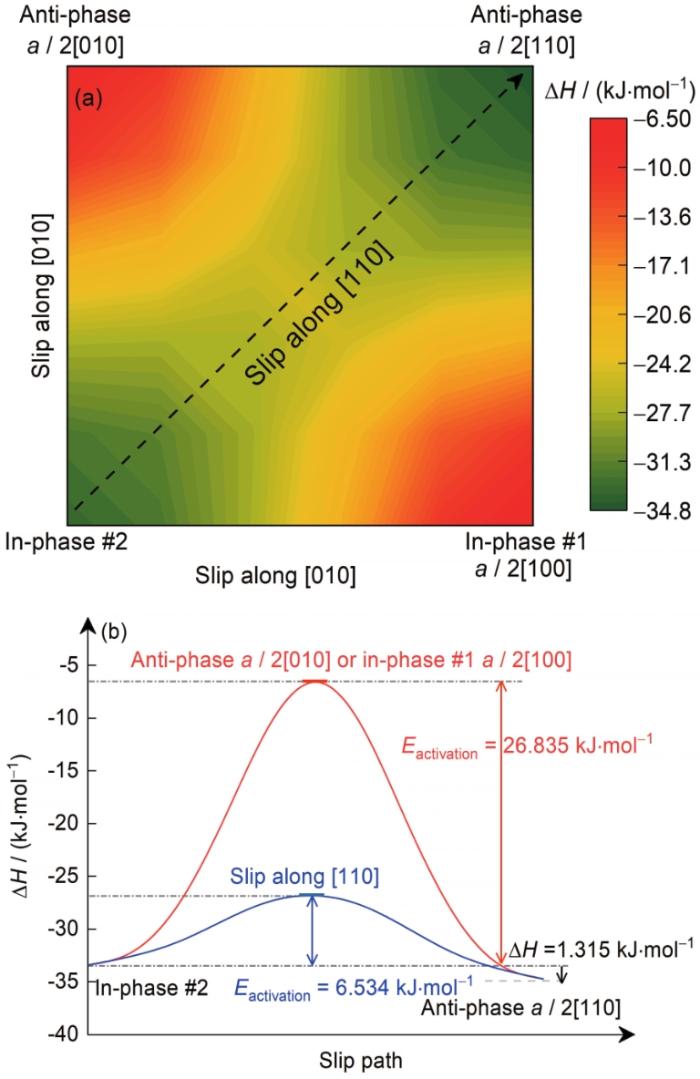
图4
δ'/θ'/δ'复合沉淀相由同相#2结构转变为反相结构的滑移势能面以及相应的滑移激活能
Fig.4
Surface free energy evolution of the δ'/θ'/δ' composite precipitate slipping from in-phase #2 to anti-phase (a), and the corresponding activ-ation energy (Eactivation) along slipping path (b)
图5
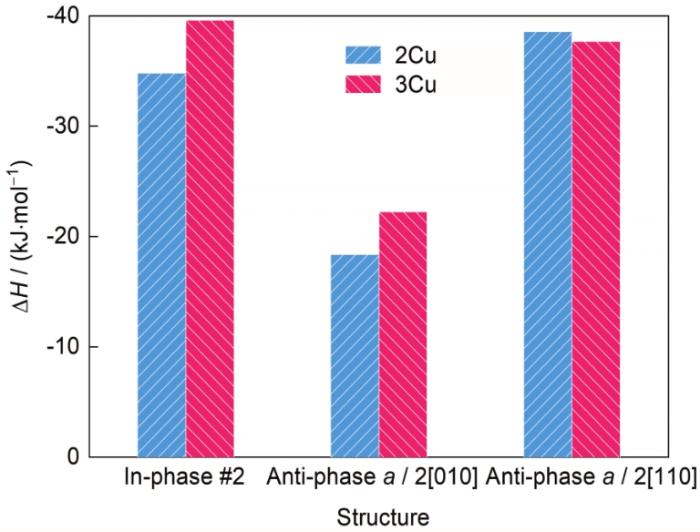
图5
包含2、3层Cu的δ'/θ'/δ'复合沉淀相采取不同位相关系时的ΔH
Fig.5
ΔH of δ'/θ'/δ' composite precipitate containing 2 and 3 Cu-layers with different relationships for the opposite δ'
2.2 不同界面结构的界面能
为了从形核热力学角度阐述不同位相结构导致的δ'相在θ'相上形核、生长的差异,计算了上述3种位相关系对应的界面结构的界面能。然而,在求解θ'/δ'两相界面能时,采用传统的线性拟合法(linear fitting)[25]、直接法(direct calculation)[26]都难以满足计算要求。如,采用线性拟合法时,界面能的计算结果会是界面过渡区的平均值;而直接法将造成能量与化学计量比的不一致性[19]。为了精确求解θ'/δ'界面能,构建了如图6所示的界面模型。图6a是θ'/δ'界面结构,将其按相种类划分出θ'和δ'相,分别得到图6b和c。需要说明的是,两相界面距离是通过构建不加真空层的块体结构,弛豫晶格参数、原子位置得到的。界面能(γinterface)采取如下公式计算[19]:
式中,A为表面面积,Esurface为表面结构的能量,Ebulk为独立相结构的能量。考虑到切开各相不符合化学计量比,ni 为多出的原子个数,μi 为多出原子对应的化学势。不同位相结构的界面能结果如表1所示。界面能由高到低依次为:反相a /
图6
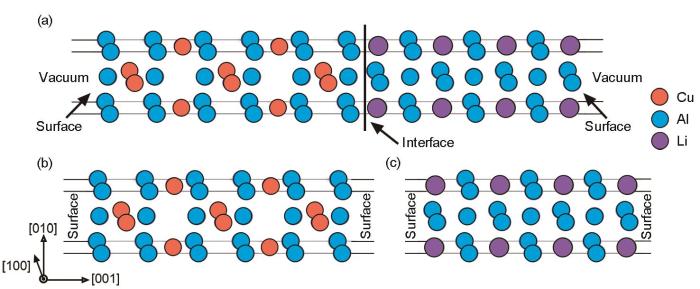
图6
求解θ'/δ'界面结构界面能的示意图
Fig.6
Schematics how solving for interfacial energy of θ'/δ' interfacial structures, the interfacial structures of θ'/δ' containing vacuum (a), separated θ' (b) and δ' (c) phases with surface
表1 3种界面结构的界面面积、界面能以及解理面各相的表面能
Table 1
| Structure | S / nm2 | |||
|---|---|---|---|---|
| In-phase #2 | 0.165 | 1.401 | 0.894 | 0.335 |
| Anti-phase a / 2[010] | 0.163 | 1.418 | 0.894 | 0.964 |
| Anti-phase a / 2[110] | 0.163 | 1.445 | 0.846 | -0.015 |
2.3 不同界面结构的理想断裂强度
图7

图7
3种θ'/δ'界面结构的键能-位移曲线以及对应的解理应力变化曲线
Fig.7
Bonding energy (Eb)-displacement (d) curves for three θ'/δ' interface structures (a) and corres-ponding cleavage stress variation curves (b)
表2 基于Rose断裂模型拟合得到的3种界面结构的临界位移(dic),解理功(Gc)以及理想解理应力(σic)
Table 2
| Structure | dic / nm | Gc / (J·m-2) | σic / GPa |
|---|---|---|---|
| In-phase #2 | 0.059 | 1.948 | 12.356 |
| Anti-phase a / 2[010] | 0.050 | 1.341 | 9.796 |
| Anti-phase a / 2[110] | 0.061 | 2.097 | 12.659 |
2.4 3种界面结构的成键分析
以包含2层Cu的δ'/θ'/δ'复合沉淀为例,进一步从化学键的成键角度分析界面间原子对成键对复合沉淀相结构稳定性的影响。图8展示了3种θ'/δ'界面结构,界面间不同原子对的成键示意图。可知,在这3种界面上主要存在2类Li—Al键,包括Li1—Al2和Li1—Al3;以及2类Al—Al键,包括Al1—Al2和Al1—Al3。表3和4分别总结了这些原子对的键长(b')以及相应的基于Hamilton矩阵的晶体轨道布居理论的原子对成键的积分值(-ICOHP)。对比不同原子对成键的-ICOHP可以得知,Al—Al键的成键强度要远大于Li—Al键,表明Al—Al键对界面结合、稳定界面结构起主导贡献。分析不同原子对的b'值得知,与同相#2和反相a / 2[110]不同的是,在反相a / 2[010]结构中,不在同一水平层(沿着δ'/θ'/δ'复合沉淀的(010)面)的原子对,包括Li1—Al2和Al1—Al3,其b'明显增加,增长约0.1 nm。而对于处于同一水平层的Li1—Al3和Al1—Al2,b'略有缩短,约0.020 nm。结合这些原子对的-ICOHP可以发现,对于键长拉伸的Li1—Al2和Al1—Al3,成键态积分值大幅降低,且只有其在同相#2和反相a /
图8
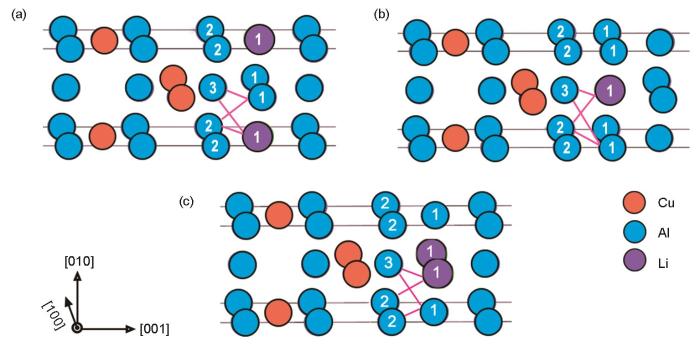
图8
位于3种θ'/δ'界面处不同原子间的成键示意图
Fig.8
Schematics of different atomic bonding in three types of θ'/δ' interfaces, including in-phase #2 (a), anti-phase a /
表3 位于3种θ′/δ′界面处,不同原子对化学键的键长 (nm)
Table 3
| Structure | Li1—Al2 | Li1—Al3 | Al1—Al2 | Al1—Al3 |
|---|---|---|---|---|
| In-phase #2 | 0.288 | 0.288 | 0.288 | 0.288 |
| Anti-phase a / 2[010] | 0.386 | 0.271 | 0.262 | 0.397 |
| Anti-phase a / 2[110] | 0.280 | 0.280 | 0.279 | 0.279 |
表4 基于Hamilton矩阵的晶体轨道布局理论,位于3种θ'/δ'界面结构中,不同原子对的积分值(-ICOHP) (eV)
Table 4
| Structure | Li1—Al2 | Li1—Al3 | Al1—Al2 | Al1—Al3 |
|---|---|---|---|---|
| In-phase #2 | 0.256 | 0.256 | 1.655 | 1.655 |
| Anti-phase a / 2[010] | 0.052 | 0.515 | 3.265 | 0.178 |
| Anti-phase a / 2[110] | 0.264 | 0.265 | 2.031 | 2.031 |
进一步分析同相#2和反相a /
将Al原子投影到3s、3p轨道,Li原子投影到1s、2s轨道,进一步分析稳定反相a /
图 9
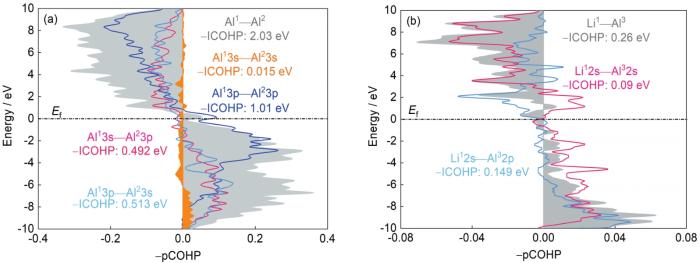
图 9
基于Hamilton举证的晶体轨道布局理论,在反相a /
Fig.9
Different orbital-pair contributions to the total Al1—Al2 (a) and Li1—Al3 (b) interactions based on the crystal orbital Hamilton population analyses for the anti-phase a /
3 结论
(1) 对于复合沉淀相δ'/θ'/δ',当θ'相包含奇数Cu层时,δ'/θ'采取反相a /
(2) 当δ'/θ'/δ'相包含奇数(偶数)层Cu原子时,δ'/θ'采取同相(反相a /
(3) 基于Rose界面断裂模型,对于包含奇数(偶数)层Cu原子的δ'/θ'/δ'相,同相#2 (反相a /
(4) 界面处Al原子与邻近层Al和Li原子间高的成键强度决定了反相a /
参考文献
Progress in characterization methods for thermoplastic deforming constitutive models of Al-Li alloys: A review
[J].
The influence of Cu/Li ratio on precipitation in Al-Cu-Li-x alloys
[J].
Microstructure and hot deformation behavior of a new aluminum-lithium-copper based AA2070 alloy
[J].
Effect of Zn additions on precipitation during aging of alloy 8090
[J].
Overview of the effects of impurities and rare earth elements in Al-Li alloys
[J].
On through thickness crystallographic texture gradient in Al-Li-Cu-Zr alloy
[J].
Anisotropic yielding stress of 2198 Al-Li alloy sheet and mechanisms
[J].
Continuous dynamic recrystallization in an Al-Li-Mg-Sc alloy during equal-channel angular extrusion
[J].
The effect of an electric field on the mechanical properties and microstructure of Al-Li alloy containing Ce
[J].
Aging kinetics of friction stir welded Al-Cu-Li-Mg-Ag and Al-Cu-Li-Mg alloys
[J].
Composite precipitates in a commercial Al-Li-Cu-Mg-Zr alloy
[J].
Influence of Mg and Li content on the microstructure evolution of Al-Cu-Li alloys during long-term ageing
[J].
Influence of Mg, Ag and Zn minor solute additions on the precipitation kinetics and strengthening of an Al-Cu-Li alloy
[J].
First-principles study of the nucleation and stability of ordered precipitates in ternary Al-Sc-Li alloys
[J].
Structures and properties of nano-precipitates in Al-Li alloys
[J].
铝锂合金纳米析出相结构与性能综述
[J].
Heterophase interface dominated deformation and mechanical properties in Al-Cu-Li Alloys
[J].
Interfacial structure evolution of the growing composite precipitates in Al-Cu-Li alloys
[J].
First-principle investigation on the interfacial structure evolution of the δ'/θ'/δ' composite precipitates in Al-Cu-Li alloys
[J].
Uncovering the influence of Cu on the thickening and strength of the δ'/θ'/δ' nano-composite precipitate in Al-Cu-Li alloys
[J].
Universal features of bonding in metal
[J].
Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids
[J].
Quantum-chemical computations of solids benefit enormously from numerically efficient plane-wave (PW) basis sets, and together with the projector augmented-wave (PAW) method, the latter have risen to one of the predominant standards in computational solid-state sciences. Despite their advantages, plane waves lack local information, which makes the interpretation of local densities-of-states (DOS) difficult and precludes the direct use of atom-resolved chemical bonding indicators such as the crystal orbital overlap population (COOP) and the crystal orbital Hamilton population (COHP) techniques. Recently, a number of methods have been proposed to overcome this fundamental issue, built around the concept of basis-set projection onto a local auxiliary basis. In this work, we propose a novel computational technique toward this goal by transferring the PW/PAW wavefunctions to a properly chosen local basis using analytically derived expressions. In particular, we describe a general approach to project both PW and PAW eigenstates onto given custom orbitals, which we then exemplify at the hand of contracted multiple-ζ Slater-type orbitals. The validity of the method presented here is illustrated by applications to chemical textbook examples-diamond, gallium arsenide, the transition-metal titanium-as well as nanoscale allotropes of carbon: a nanotube and the C60 fullerene. Remarkably, the analytical approach not only recovers the total and projected electronic DOS with a high degree of confidence, but it also yields a realistic chemical-bonding picture in the framework of the projected COHP method.Copyright © 2013 Wiley Periodicals, Inc.
Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set
[J].
Special points for Brillouin-zone integrations
[J].
Multiscale modeling of θ′ precipitation in Al-Cu binary alloys
[J].
First-principles calculations of β″-Mg5Si6/α-Al interfaces
[J].
Designing interfaces in energy materials applications with first-principles calculations
[J].
ADAIS: Automatic derivation of anisotropic ideal strength via high-throughput first-principles computations
[J].Anisotropic ideal strength is a fundamental and important plasticity parameter in scaling the intrinsic strength of strong crystalline materials, and is a potential descriptor in searching and designing novel hard/superhard materials. However, to the best of our knowledge, an automatic derivation of anisotropic ideal strength has not been implemented in any open-source code available so far. In this paper, we present our developed ADAIS code, an automatic derivation of anisotropic ideal strength via high-throughput first-principles computations for both three-dimensional and two-dimensional crystalline materials with any symmetry, as well as for an ideal interface model. Several fundamental mechanical quantities can be automatically derived, including ideal tensile and shear strengths through affine deformation, universal binding energy and generalized stacking fault energy, as well as the ideal cleavage and slide stresses through alias deformation. The implementation of this code has been comprehensively demonstrated and critically validated by a lot of evaluations and tests of various crystalline materials with different symmetry, indicating that our code could provide a high-efficiency solution to quantify the strength of strong solids. (C) 2018 Elsevier B.V.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}