低合金钢TMCP中相变热力学/动力学相关性探讨
3
2016
... 然而,析出相研究往往受限于实验技术:基体中弥散分布的纳米级微粒的实时原位观测仍很困难;多相竞争析出时,过渡亚稳相因短暂出现而难以通过实验测得;同成分析出相因具有多种非常接近的晶体结构,而难以表征区分;析出相形状和成分变化进一步增加实验测量难度.诸如此类,实验上的困难重重促使理论模拟成为探索析出相潜在形成机理的不二途径.体系与环境间的交互作用(例如功、热和物质传输)由热力学第一定律描述,而第二定律主导体系状态的演化,即热力学稳定性的变化.改变成分与工艺可调控析出反应的热力学驱动力和动力学能垒,相变热-动力学协同控制析出相形成机理及最终演化状态,因此,模拟相变重在精确表征热-动力学.本团队研究表明,相变热-动力学相互关联,热力学驱动力和动力学能垒间存在互斥关系[1 ,2 ] ,即热力学表征精度会直接影响动力学模拟结果.不同的模拟方法区别在于对析出热-动力学协同作用描述的尺度、角度与精度,因而各具特色,良莠不齐.本文旨在综述前人在析出相模拟方面的代表性工作,助力与此相关的金属材料理性设计. ...
... 形核阶段晶核细小,原子扩散距离短,析出由界面反应控制;随晶核变大,基体过饱和度不断下降,析出驱动力减小,长大动力学由扩散主导.这完美地反映出热力学驱动力与动力学能垒的互斥关系[1 ,2 ] .Zener[17 ] 给出球形晶核在小过饱和度(c 0 -c i ≪c p -χ i )和局域平衡假设下长大速率v : ...
... 数据库构建是材料基因工程的关键要素,但是,目前的理性设计通过人工智能的机器学习技术找到“成分-工艺-组织-性能”的对应关系,仍存在很大改进空间.一方面,缺乏足够的数据来训练出准确的模型;另一方面,机器学习是个黑箱操作过程,物理意义不明确,无法做深层次的相变机理研究.因此,基于热-动力学协同的析出相模拟显得至关重要.相变中的热-动力学协同作用表现为热-动力学相关性[1 ,2 ] ,若将其纳入模型中,既可以使符合这一普遍科学规律的模拟结果更加准确,也有助于探索析出机理.本文重在阐述获取成分-工艺-析出组织间映射关系的方法,而理性设计的最终目的为获取材料优异性能. ...
低合金钢TMCP中相变热力学/动力学相关性探讨
3
2016
... 然而,析出相研究往往受限于实验技术:基体中弥散分布的纳米级微粒的实时原位观测仍很困难;多相竞争析出时,过渡亚稳相因短暂出现而难以通过实验测得;同成分析出相因具有多种非常接近的晶体结构,而难以表征区分;析出相形状和成分变化进一步增加实验测量难度.诸如此类,实验上的困难重重促使理论模拟成为探索析出相潜在形成机理的不二途径.体系与环境间的交互作用(例如功、热和物质传输)由热力学第一定律描述,而第二定律主导体系状态的演化,即热力学稳定性的变化.改变成分与工艺可调控析出反应的热力学驱动力和动力学能垒,相变热-动力学协同控制析出相形成机理及最终演化状态,因此,模拟相变重在精确表征热-动力学.本团队研究表明,相变热-动力学相互关联,热力学驱动力和动力学能垒间存在互斥关系[1 ,2 ] ,即热力学表征精度会直接影响动力学模拟结果.不同的模拟方法区别在于对析出热-动力学协同作用描述的尺度、角度与精度,因而各具特色,良莠不齐.本文旨在综述前人在析出相模拟方面的代表性工作,助力与此相关的金属材料理性设计. ...
... 形核阶段晶核细小,原子扩散距离短,析出由界面反应控制;随晶核变大,基体过饱和度不断下降,析出驱动力减小,长大动力学由扩散主导.这完美地反映出热力学驱动力与动力学能垒的互斥关系[1 ,2 ] .Zener[17 ] 给出球形晶核在小过饱和度(c 0 -c i ≪c p -χ i )和局域平衡假设下长大速率v : ...
... 数据库构建是材料基因工程的关键要素,但是,目前的理性设计通过人工智能的机器学习技术找到“成分-工艺-组织-性能”的对应关系,仍存在很大改进空间.一方面,缺乏足够的数据来训练出准确的模型;另一方面,机器学习是个黑箱操作过程,物理意义不明确,无法做深层次的相变机理研究.因此,基于热-动力学协同的析出相模拟显得至关重要.相变中的热-动力学协同作用表现为热-动力学相关性[1 ,2 ] ,若将其纳入模型中,既可以使符合这一普遍科学规律的模拟结果更加准确,也有助于探索析出机理.本文重在阐述获取成分-工艺-析出组织间映射关系的方法,而理性设计的最终目的为获取材料优异性能. ...
Martensitic transition in Fe via Bain path at finite temperatures: A comprehensive first-principles study
3
2018
... 然而,析出相研究往往受限于实验技术:基体中弥散分布的纳米级微粒的实时原位观测仍很困难;多相竞争析出时,过渡亚稳相因短暂出现而难以通过实验测得;同成分析出相因具有多种非常接近的晶体结构,而难以表征区分;析出相形状和成分变化进一步增加实验测量难度.诸如此类,实验上的困难重重促使理论模拟成为探索析出相潜在形成机理的不二途径.体系与环境间的交互作用(例如功、热和物质传输)由热力学第一定律描述,而第二定律主导体系状态的演化,即热力学稳定性的变化.改变成分与工艺可调控析出反应的热力学驱动力和动力学能垒,相变热-动力学协同控制析出相形成机理及最终演化状态,因此,模拟相变重在精确表征热-动力学.本团队研究表明,相变热-动力学相互关联,热力学驱动力和动力学能垒间存在互斥关系[1 ,2 ] ,即热力学表征精度会直接影响动力学模拟结果.不同的模拟方法区别在于对析出热-动力学协同作用描述的尺度、角度与精度,因而各具特色,良莠不齐.本文旨在综述前人在析出相模拟方面的代表性工作,助力与此相关的金属材料理性设计. ...
... 形核阶段晶核细小,原子扩散距离短,析出由界面反应控制;随晶核变大,基体过饱和度不断下降,析出驱动力减小,长大动力学由扩散主导.这完美地反映出热力学驱动力与动力学能垒的互斥关系[1 ,2 ] .Zener[17 ] 给出球形晶核在小过饱和度(c 0 -c i ≪c p -χ i )和局域平衡假设下长大速率v : ...
... 数据库构建是材料基因工程的关键要素,但是,目前的理性设计通过人工智能的机器学习技术找到“成分-工艺-组织-性能”的对应关系,仍存在很大改进空间.一方面,缺乏足够的数据来训练出准确的模型;另一方面,机器学习是个黑箱操作过程,物理意义不明确,无法做深层次的相变机理研究.因此,基于热-动力学协同的析出相模拟显得至关重要.相变中的热-动力学协同作用表现为热-动力学相关性[1 ,2 ] ,若将其纳入模型中,既可以使符合这一普遍科学规律的模拟结果更加准确,也有助于探索析出机理.本文重在阐述获取成分-工艺-析出组织间映射关系的方法,而理性设计的最终目的为获取材料优异性能. ...
Ocean of data: Integrating first-principles calculations and CALPHAD modeling with machine learning
1
2018
... CALPHAD (CALculation PHAse Diagram)[3 ] 计算热力学已被广泛应用于数据库构建,多组分相平衡计算及材料特性和温度、压力与组分间的函数关系获取.然而传统CALPHAD通常依赖于实验数据,亚稳相数据缺乏,加上模拟精度受实验误差影响,故数据库的更新会导致模拟结果的显著变化[4 ] .基于密度泛函理论[5 ,6 ] 的第一性原理计算,只需给定原子种类与晶体结构,便可通过描述原子和电子层次交互作用来近似求解Schrodinger方程以获得体系热力学数据.此外,在材料基因工程[7 ,8 ] 背景下,计算材料学与材料信息学交汇融合,人工智能技术为从第一性原理计算出发的热力学数据库构建创造了可能. ...
Thermodynamic prediction of martensitic transformation temperature in Fe-Ni-C system
1
2020
... CALPHAD (CALculation PHAse Diagram)[3 ] 计算热力学已被广泛应用于数据库构建,多组分相平衡计算及材料特性和温度、压力与组分间的函数关系获取.然而传统CALPHAD通常依赖于实验数据,亚稳相数据缺乏,加上模拟精度受实验误差影响,故数据库的更新会导致模拟结果的显著变化[4 ] .基于密度泛函理论[5 ,6 ] 的第一性原理计算,只需给定原子种类与晶体结构,便可通过描述原子和电子层次交互作用来近似求解Schrodinger方程以获得体系热力学数据.此外,在材料基因工程[7 ,8 ] 背景下,计算材料学与材料信息学交汇融合,人工智能技术为从第一性原理计算出发的热力学数据库构建创造了可能. ...
Inhomogeneous electron gas
1
1964
... CALPHAD (CALculation PHAse Diagram)[3 ] 计算热力学已被广泛应用于数据库构建,多组分相平衡计算及材料特性和温度、压力与组分间的函数关系获取.然而传统CALPHAD通常依赖于实验数据,亚稳相数据缺乏,加上模拟精度受实验误差影响,故数据库的更新会导致模拟结果的显著变化[4 ] .基于密度泛函理论[5 ,6 ] 的第一性原理计算,只需给定原子种类与晶体结构,便可通过描述原子和电子层次交互作用来近似求解Schrodinger方程以获得体系热力学数据.此外,在材料基因工程[7 ,8 ] 背景下,计算材料学与材料信息学交汇融合,人工智能技术为从第一性原理计算出发的热力学数据库构建创造了可能. ...
Self-consistent equations including exchange and correlation effects
1
1965
... CALPHAD (CALculation PHAse Diagram)[3 ] 计算热力学已被广泛应用于数据库构建,多组分相平衡计算及材料特性和温度、压力与组分间的函数关系获取.然而传统CALPHAD通常依赖于实验数据,亚稳相数据缺乏,加上模拟精度受实验误差影响,故数据库的更新会导致模拟结果的显著变化[4 ] .基于密度泛函理论[5 ,6 ] 的第一性原理计算,只需给定原子种类与晶体结构,便可通过描述原子和电子层次交互作用来近似求解Schrodinger方程以获得体系热力学数据.此外,在材料基因工程[7 ,8 ] 背景下,计算材料学与材料信息学交汇融合,人工智能技术为从第一性原理计算出发的热力学数据库构建创造了可能. ...
The materials genome initiative, the interplay of experiment, theory and computation
1
2014
... CALPHAD (CALculation PHAse Diagram)[3 ] 计算热力学已被广泛应用于数据库构建,多组分相平衡计算及材料特性和温度、压力与组分间的函数关系获取.然而传统CALPHAD通常依赖于实验数据,亚稳相数据缺乏,加上模拟精度受实验误差影响,故数据库的更新会导致模拟结果的显著变化[4 ] .基于密度泛函理论[5 ,6 ] 的第一性原理计算,只需给定原子种类与晶体结构,便可通过描述原子和电子层次交互作用来近似求解Schrodinger方程以获得体系热力学数据.此外,在材料基因工程[7 ,8 ] 背景下,计算材料学与材料信息学交汇融合,人工智能技术为从第一性原理计算出发的热力学数据库构建创造了可能. ...
New frontiers for the materials genome initiative
1
2019
... CALPHAD (CALculation PHAse Diagram)[3 ] 计算热力学已被广泛应用于数据库构建,多组分相平衡计算及材料特性和温度、压力与组分间的函数关系获取.然而传统CALPHAD通常依赖于实验数据,亚稳相数据缺乏,加上模拟精度受实验误差影响,故数据库的更新会导致模拟结果的显著变化[4 ] .基于密度泛函理论[5 ,6 ] 的第一性原理计算,只需给定原子种类与晶体结构,便可通过描述原子和电子层次交互作用来近似求解Schrodinger方程以获得体系热力学数据.此外,在材料基因工程[7 ,8 ] 背景下,计算材料学与材料信息学交汇融合,人工智能技术为从第一性原理计算出发的热力学数据库构建创造了可能. ...
Homogeneous second-phase precipitation
4
2001
... 目前,析出相形成热-动力学机理研究已很广泛,将合金在高温单相区均匀化处理,随后缓冷至两相区,或快冷再升温至两相区进行时效,均可引起析出反应;新相形成需经历形核、长大、粗化阶段[9 ] ,且同时出现在基体的各个微区;形核长大驱动力源于新旧两相间自由能差,粗化驱动力源自界面能的下降.不同方法在预测析出物状态(结构、形貌、成分、尺寸分布、体积分数、数密度)时,都需依据形核、长大、粗化的客观事实.本节简单回顾形核、长大、粗化的经典理论,同时简介处理非平衡复杂多组分多相体系中扩散的有效工具,即热力学极值原理[10 ] ,作为后文模拟的理论基础. ...
... 要获得平均尺寸演化,需知临界尺寸微粒数量,引入临界尺寸微粒处的密度f a 为[9 ] : ...
... 式中,c ¯ 25 )、(27 )~(29 )联立即可描述平均尺寸及数量演化.然而,LS理论中求解界面成分时采用线性形式的Gibbs-Thomson方程,不适用于小微粒,后来Kampmann和Wagner通过引入非线性Gibbs-Thomson方程(式(8) )修正了LS模型,即著名的MLS (modified Langer-Schwartz)理论[9 ,46 ,47 ] .平均尺寸法虽计算量小,但无法描述微粒尺寸分布[48 ] ,而该参量可借助尺寸分级法获得. ...
... (1) Kampmann-Wagner数值方法.Wagner等[9 ] 在MLS模型基础上提出了求解连续性方程式(23) 的数值方法,可以通过同时求解式(23) 和平均场近似下的质量守恒方程式(30) 来直接得到微粒尺寸分布f (R )的演化,此即为KWN (Kampmann-Wagner numerical)模型. ...
Application of the thermodynamic extremal principle to modeling of thermodynamic processes in material sciences
1
2005
... 目前,析出相形成热-动力学机理研究已很广泛,将合金在高温单相区均匀化处理,随后缓冷至两相区,或快冷再升温至两相区进行时效,均可引起析出反应;新相形成需经历形核、长大、粗化阶段[9 ] ,且同时出现在基体的各个微区;形核长大驱动力源于新旧两相间自由能差,粗化驱动力源自界面能的下降.不同方法在预测析出物状态(结构、形貌、成分、尺寸分布、体积分数、数密度)时,都需依据形核、长大、粗化的客观事实.本节简单回顾形核、长大、粗化的经典理论,同时简介处理非平衡复杂多组分多相体系中扩散的有效工具,即热力学极值原理[10 ] ,作为后文模拟的理论基础. ...
Implementation of classical nucleation and growth theories for precipitation
7
2008
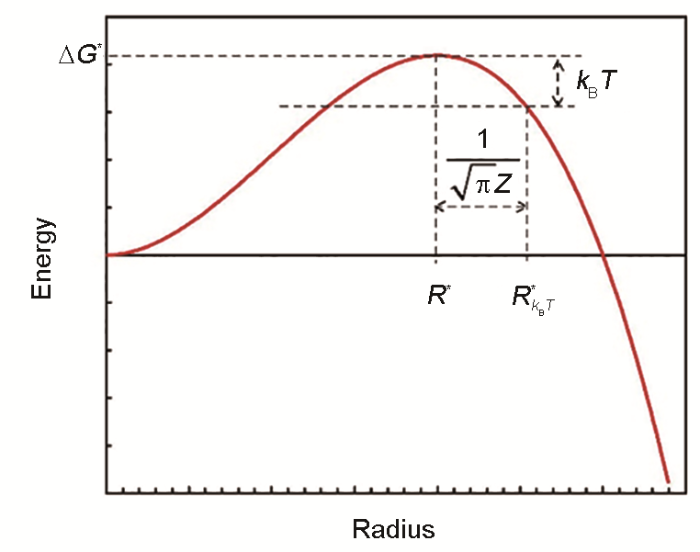
... 以描述相变体系自由能变化为基础,经典形核理论针对稳定核胚形成的动力学问题.如图1 [11 ] 所示,对于一个具有半径R 的球形析出相的形成,其体系自由能变化ΔG (R )可以表示为: ...
... [
11 ]
Schematic representation of the Gibbs energy changes associated with precipitate formation as a function of their radius <i>R</i> in the classical nucleation theory (Δ<i>G</i><sup>*</sup> is the nucleation barrier, <i>R</i><sup>*</sup> is the critical radius for stable precipitates, <span class="formulaText"><inline-formula><math id="M1"><msubsup><mrow><mi>R</mi></mrow><mrow><msub><mrow><mi>k</mi></mrow><mrow><mi mathvariant="normal">B</mi></mrow></msub><mi>T</mi></mrow><mrow><mi mathvariant="normal">*</mi></mrow></msubsup></math></span></inline-formula></span> is the radius at which stable precipitates nucleate, <i>Z</i> is the Zeldovich factor, <i>k</i><sub>B</sub> is the Boltzmann constant and <i>T</i> is the temperature)<sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup> Fig.1 ![]()
Δ G ( R ) = 4 3 π R 3 Δ g + 4 π R 2 γ (1) 式中,Δg 是单位体积析出驱动力(化学自由能Δg chem 和应变能Δg s 总和),γ 是界面能.90多年前,Volmer和Weber[12 ] 指出形核速率J 遵循以下形式: ...
... [
11 ]
Fig.1 ![]()
Δ G ( R ) = 4 3 π R 3 Δ g + 4 π R 2 γ (1) 式中,Δg 是单位体积析出驱动力(化学自由能Δg chem 和应变能Δg s 总和),γ 是界面能.90多年前,Volmer和Weber[12 ] 指出形核速率J 遵循以下形式: ...
... 微观结构模拟方法可划分为直接精细数值(direct detailed numerical)法和频率分布函数(frequency distribution function)法[44 ] .前者以相场法为代表;后者通常指基于经典形核-长大-粗化理论的模拟,通过对结构特征的统计处理来完整地描述析出全过程.在频率分布函数法中,可依据形核长大速率方程的应用方式进一步划分为平均尺寸法与尺寸分级法[11 ] ,此为本节重点. ...
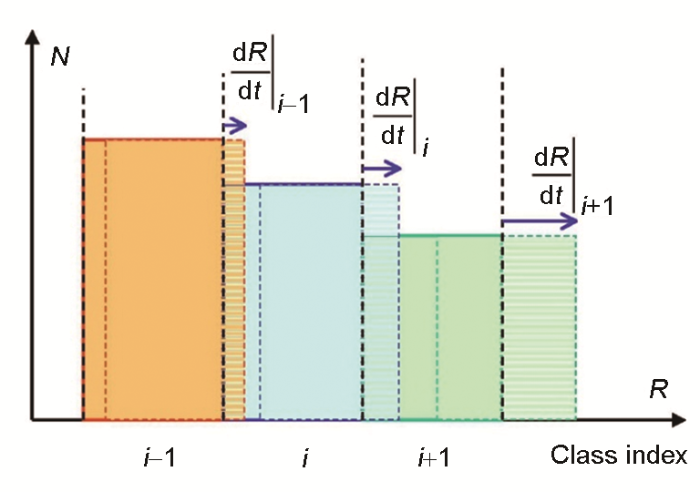
... 式中,C 0 为初始基体溶质浓度.该方法开创了尺寸分级法的先河,通过将图4 的微粒尺寸分布f (R )离散为许多部分,即划分尺寸等级,然后将尺寸的连续时间演化离散为单个时间步进行.KWN模型随后被不断改进,Myhr和Grong[49 ] 提出了计算相邻尺寸等级之间通量的有限差分方法,如图5 [11 ] 所示,也称作“类Euler法”.Nicolas和Deschamps[50 ] 将KWN模型应用到非等温过程中.Du等[51 ] 改善了长大速率方程,考虑高过饱和度情形和交叉扩散后,通过求解界面移动速率、变化的tie-line和物质流守恒方程使得KWN模型可得到多组分析出物的成分变化.由于实际晶核往往是非球形的[52 ~54 ] ,不同形状晶核因伴随不同的应变能和界面能变化而具有不同的析出过程,模拟需基于晶核的真实形貌.于是Holmedal等[55 ] 和Li等[44 ] 通过引入长大速率和Gibbs-Thomson效应的修正因子将KWN模型用于处理非球形析出相. ...
... [
11 ]
Growth of precipitates in the “Euler-like approach” (At each time step, fluxes between each neighboring classes are calculated. <i>N</i> is the number of particles in a specific size class, and <span class="formulaText"><inline-formula><math id="M59"><mfrac><mrow><mi mathvariant="normal">d</mi><mi>R</mi></mrow><mrow><mi mathvariant="normal">d</mi><mi>t</mi></mrow></mfrac></math></span></inline-formula></span> is the growth rate)<sup>[<xref ref-type="bibr" rid="R11">11</xref>]</sup> Fig.5 ![]()
KWN模型已成为Thermal-Calc软件中TC-PRISMA模块的核心理论,被广泛应用于复杂工业合金的析出相模拟、析出机理探究与经典理论发展.微粒尺寸分布,即一定尺寸范围内的析出物数量,对金属性能有很大影响.Robson[48 ] 借助KWN模型模拟了Cu-1.02%Co (原子分数)合金600℃下的析出反应,结果表明早期存在着晶核尺寸的双峰分布,是由形核与长大速率变化引起的,该现象也在其他实验与模拟中发现[49 ,56 ~58 ] .Robson[59 ] 还研究了形核、长大、粗化阶段出现的重叠现象,结果表明重叠程度与界面能、溶质过饱和度有很大关联而与溶质扩散性无关. ...
... [
11 ]
Fig.5 ![]()
KWN模型已成为Thermal-Calc软件中TC-PRISMA模块的核心理论,被广泛应用于复杂工业合金的析出相模拟、析出机理探究与经典理论发展.微粒尺寸分布,即一定尺寸范围内的析出物数量,对金属性能有很大影响.Robson[48 ] 借助KWN模型模拟了Cu-1.02%Co (原子分数)合金600℃下的析出反应,结果表明早期存在着晶核尺寸的双峰分布,是由形核与长大速率变化引起的,该现象也在其他实验与模拟中发现[49 ,56 ~58 ] .Robson[59 ] 还研究了形核、长大、粗化阶段出现的重叠现象,结果表明重叠程度与界面能、溶质过饱和度有很大关联而与溶质扩散性无关. ...
Keimbildung in übers?ttigten gebilden
1
1926
... 式中,Δg 是单位体积析出驱动力(化学自由能Δg chem 和应变能Δg s 总和),γ 是界面能.90多年前,Volmer和Weber[12 ] 指出形核速率J 遵循以下形式: ...
Kinetische behandlung der keimbildung in übers?ttigten d?mpfen
1
1935
... 式中,ΔG * 是形成半径为R * (R * 为稳定析出相的临界半径)的临界核胚所需的能量,k B 是Boltzmann常数,T 是温度.随后Becker和Dӧring[13 ] 与Zeldvich[14 ] 给出式(2) 中指前因子并得到稳态形核速率J ss : ...
On the theory of new phase formation: Cavitation
1
1943
... 式中,ΔG * 是形成半径为R * (R * 为稳定析出相的临界半径)的临界核胚所需的能量,k B 是Boltzmann常数,T 是温度.随后Becker和Dӧring[13 ] 与Zeldvich[14 ] 给出式(2) 中指前因子并得到稳态形核速率J ss : ...
1
1968
... 式中,v P 是平均原子体积.对于球形微粒,Russel[15 ] 给出β * 表达式: ...
K
1
1984
... 式中,D 是原子扩散系数,x 是母相平均原子分数,a 是晶格参数.由于式(3) 并不提供微粒尺寸分布达到稳态前的动力学信息,Kampmann和Wagner[16 ] 引入孕育期τ ,给出与时间t 相关的形核速率J * : ...
Theory of growth of spherical precipitates from solid solution
1
1949
... 形核阶段晶核细小,原子扩散距离短,析出由界面反应控制;随晶核变大,基体过饱和度不断下降,析出驱动力减小,长大动力学由扩散主导.这完美地反映出热力学驱动力与动力学能垒的互斥关系[1 ,2 ] .Zener[17 ] 给出球形晶核在小过饱和度(c 0 -c i ≪c p -χ i )和局域平衡假设下长大速率v : ...
Gibbs-Thomson effects in phase transformations
1
2005
... 式中,c 0 为基体溶质浓度,c i 为界面处基体平衡溶质浓度,c p 为析出相溶质浓度.由于Gibbs-Thomson效应[18 ] ,界面曲率依下式影响c i : ...
The theory of Ostwald ripening
1
1985
... 式(8) 表明,小晶核的c i 比大微粒大,在析出后期,当c 0 足够小时,式(7) 中(c 0 -c i )项对于小晶核会小于0,而对于大晶核会大于0,即发生前者溶解后者长大的粗化现象,亦称“Ostwald熟化”[19 ] .该现象可由Lifchitz和Slyosov[20 ] 与Wagner[21 ] 先后发展的经典LSW (Lifchitz-Slyosov-Wagner)粗化理论描述,他们基于下式计算了晶核尺寸分布f (R )的演化: ...
The kinetics of precipitation from supersaturated solid solutions
1
1961
... 式(8) 表明,小晶核的c i 比大微粒大,在析出后期,当c 0 足够小时,式(7) 中(c 0 -c i )项对于小晶核会小于0,而对于大晶核会大于0,即发生前者溶解后者长大的粗化现象,亦称“Ostwald熟化”[19 ] .该现象可由Lifchitz和Slyosov[20 ] 与Wagner[21 ] 先后发展的经典LSW (Lifchitz-Slyosov-Wagner)粗化理论描述,他们基于下式计算了晶核尺寸分布f (R )的演化: ...
Theorie der Alterung von Niederschl?gen durch Uml?sen (Ostwald-Reifung)
1
1961
... 式(8) 表明,小晶核的c i 比大微粒大,在析出后期,当c 0 足够小时,式(7) 中(c 0 -c i )项对于小晶核会小于0,而对于大晶核会大于0,即发生前者溶解后者长大的粗化现象,亦称“Ostwald熟化”[19 ] .该现象可由Lifchitz和Slyosov[20 ] 与Wagner[21 ] 先后发展的经典LSW (Lifchitz-Slyosov-Wagner)粗化理论描述,他们基于下式计算了晶核尺寸分布f (R )的演化: ...
Reciprocal relations in irreversible processes. I
2
1931
... 材料组织结构演化属于热力学非平衡过程,对此,可用动力学方程来描述体系内部状态变量,例如Fick第二定律描述了扩散场在时空间的演化.对于复杂体系,则需同时考虑置换、间隙原子与空位的运动,精确求解扩散场尤为困难.热力学极值原理是处理该复杂情形的有效工具,据其推导的演化控制方程因形式简单也非偏微分方程,既可弥补传统唯象模型的复杂性也能得到具有物理意义的解.该方法最早由Onsager[22 ] 在1931年构建,在30年后被Ziegler[23 ,24 ] 推广为处理非平衡热力学体系的变分原理,此后出现了很多其他变分形式,具体可见文献[25 ~27 ].本文介绍与SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型[28 ,29 ] 及本团队构建的多尺度组织预测模型[30 ] 相关的Ziegler热力学极值原理(亦称“最大熵产生原理”)在微观结构预测中的应用. ...
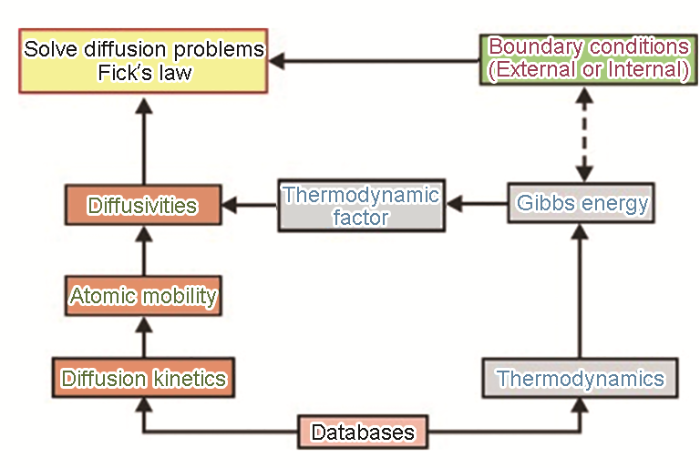
... Thermal-Calc Software AB公司发行的DICTRA软件可基于局域平衡和尖锐界面假设对多组分扩散方程求数值解,以完成复杂合金的扩散析出模拟.虽然DICTRA可处理复杂体系中扩散过程,但只适用于一维情形,即可简化成一维的几何形状(无限大且具有一定厚度的平板、无限长且具有一定大小底面的圆柱和具有一定半径的球体),模拟边界条件可根据问题灵活选择.DICTRA包含以下模块:(i) 扩散方程求解;(ii) 热力学平衡计算;(iii) 流守恒方程求解;(iv) 相界面位置移动和网格划分调整.如图2 [37 ] 所示,主要工作为求解多组分扩散方程[22 ] : ...
Some extremum principles in irreversible thermodynamics with applications to continuum mechanics
1
1963
... 材料组织结构演化属于热力学非平衡过程,对此,可用动力学方程来描述体系内部状态变量,例如Fick第二定律描述了扩散场在时空间的演化.对于复杂体系,则需同时考虑置换、间隙原子与空位的运动,精确求解扩散场尤为困难.热力学极值原理是处理该复杂情形的有效工具,据其推导的演化控制方程因形式简单也非偏微分方程,既可弥补传统唯象模型的复杂性也能得到具有物理意义的解.该方法最早由Onsager[22 ] 在1931年构建,在30年后被Ziegler[23 ,24 ] 推广为处理非平衡热力学体系的变分原理,此后出现了很多其他变分形式,具体可见文献[25 ~27 ].本文介绍与SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型[28 ,29 ] 及本团队构建的多尺度组织预测模型[30 ] 相关的Ziegler热力学极值原理(亦称“最大熵产生原理”)在微观结构预测中的应用. ...
1
1977
... 材料组织结构演化属于热力学非平衡过程,对此,可用动力学方程来描述体系内部状态变量,例如Fick第二定律描述了扩散场在时空间的演化.对于复杂体系,则需同时考虑置换、间隙原子与空位的运动,精确求解扩散场尤为困难.热力学极值原理是处理该复杂情形的有效工具,据其推导的演化控制方程因形式简单也非偏微分方程,既可弥补传统唯象模型的复杂性也能得到具有物理意义的解.该方法最早由Onsager[22 ] 在1931年构建,在30年后被Ziegler[23 ,24 ] 推广为处理非平衡热力学体系的变分原理,此后出现了很多其他变分形式,具体可见文献[25 ~27 ].本文介绍与SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型[28 ,29 ] 及本团队构建的多尺度组织预测模型[30 ] 相关的Ziegler热力学极值原理(亦称“最大熵产生原理”)在微观结构预测中的应用. ...
Thermodynamic extremal principles for irreversible processes in materials science
1
2014
... 材料组织结构演化属于热力学非平衡过程,对此,可用动力学方程来描述体系内部状态变量,例如Fick第二定律描述了扩散场在时空间的演化.对于复杂体系,则需同时考虑置换、间隙原子与空位的运动,精确求解扩散场尤为困难.热力学极值原理是处理该复杂情形的有效工具,据其推导的演化控制方程因形式简单也非偏微分方程,既可弥补传统唯象模型的复杂性也能得到具有物理意义的解.该方法最早由Onsager[22 ] 在1931年构建,在30年后被Ziegler[23 ,24 ] 推广为处理非平衡热力学体系的变分原理,此后出现了很多其他变分形式,具体可见文献[25 ~27 ].本文介绍与SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型[28 ,29 ] 及本团队构建的多尺度组织预测模型[30 ] 相关的Ziegler热力学极值原理(亦称“最大熵产生原理”)在微观结构预测中的应用. ...
On the relation between the principle of maximum dissipation and inelastic evolution given by dissipation potentials
2008
Thermodynamic treatment of diffusive phase transformation (reactive diffusion)
1
2017
... 材料组织结构演化属于热力学非平衡过程,对此,可用动力学方程来描述体系内部状态变量,例如Fick第二定律描述了扩散场在时空间的演化.对于复杂体系,则需同时考虑置换、间隙原子与空位的运动,精确求解扩散场尤为困难.热力学极值原理是处理该复杂情形的有效工具,据其推导的演化控制方程因形式简单也非偏微分方程,既可弥补传统唯象模型的复杂性也能得到具有物理意义的解.该方法最早由Onsager[22 ] 在1931年构建,在30年后被Ziegler[23 ,24 ] 推广为处理非平衡热力学体系的变分原理,此后出现了很多其他变分形式,具体可见文献[25 ~27 ].本文介绍与SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型[28 ,29 ] 及本团队构建的多尺度组织预测模型[30 ] 相关的Ziegler热力学极值原理(亦称“最大熵产生原理”)在微观结构预测中的应用. ...
Modelling of kinetics in multi-component multi-phase systems with spherical precipitates: I: Theory
3
2004
... 材料组织结构演化属于热力学非平衡过程,对此,可用动力学方程来描述体系内部状态变量,例如Fick第二定律描述了扩散场在时空间的演化.对于复杂体系,则需同时考虑置换、间隙原子与空位的运动,精确求解扩散场尤为困难.热力学极值原理是处理该复杂情形的有效工具,据其推导的演化控制方程因形式简单也非偏微分方程,既可弥补传统唯象模型的复杂性也能得到具有物理意义的解.该方法最早由Onsager[22 ] 在1931年构建,在30年后被Ziegler[23 ,24 ] 推广为处理非平衡热力学体系的变分原理,此后出现了很多其他变分形式,具体可见文献[25 ~27 ].本文介绍与SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型[28 ,29 ] 及本团队构建的多尺度组织预测模型[30 ] 相关的Ziegler热力学极值原理(亦称“最大熵产生原理”)在微观结构预测中的应用. ...
... 对于复杂合金体系,相比偏微分扩散方程,由热力学极值原理得到的演化方程求解通常更为简单.Svoboda等[28 ] 和Kozeschnik等[29 ,63 ] 推导出控制多组分多相复杂合金体系下微粒析出演化的SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型.对含有n 种组分(n =n s +n k ,n s 和n k 分别为置换和间隙原子数量)的合金体系,Ni 表示i 组元在体系中总数量(i =1, 2, ⋯ n ),Rp 表示析出相p 的半径(p =1, 2, ⋯ m ),p ci i 组元在相p 的平均浓度,γp 表示析出相p 的界面能,0 Ni 表示基体中i 组元数量,0 μi 表示i 组元在基体中化学势,p μi i 组元在相p 中化学势,则多种晶核析出时体系自由能可表示为: ...
... 式中,p c ˙ i i 组元在相p 的平均浓度变化率.求解可得控制体系演化的线性方程组,具体形式可参阅参考文献[28 ]. ...
Modelling of kinetics in multi-component multi-phase systems with spherical precipitates: II: Numerical solution and application
5
2004
... 材料组织结构演化属于热力学非平衡过程,对此,可用动力学方程来描述体系内部状态变量,例如Fick第二定律描述了扩散场在时空间的演化.对于复杂体系,则需同时考虑置换、间隙原子与空位的运动,精确求解扩散场尤为困难.热力学极值原理是处理该复杂情形的有效工具,据其推导的演化控制方程因形式简单也非偏微分方程,既可弥补传统唯象模型的复杂性也能得到具有物理意义的解.该方法最早由Onsager[22 ] 在1931年构建,在30年后被Ziegler[23 ,24 ] 推广为处理非平衡热力学体系的变分原理,此后出现了很多其他变分形式,具体可见文献[25 ~27 ].本文介绍与SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型[28 ,29 ] 及本团队构建的多尺度组织预测模型[30 ] 相关的Ziegler热力学极值原理(亦称“最大熵产生原理”)在微观结构预测中的应用. ...
... 对于复杂合金体系,相比偏微分扩散方程,由热力学极值原理得到的演化方程求解通常更为简单.Svoboda等[28 ] 和Kozeschnik等[29 ,63 ] 推导出控制多组分多相复杂合金体系下微粒析出演化的SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型.对含有n 种组分(n =n s +n k ,n s 和n k 分别为置换和间隙原子数量)的合金体系,Ni 表示i 组元在体系中总数量(i =1, 2, ⋯ n ),Rp 表示析出相p 的半径(p =1, 2, ⋯ m ),p ci i 组元在相p 的平均浓度,γp 表示析出相p 的界面能,0 Ni 表示基体中i 组元数量,0 μi 表示i 组元在基体中化学势,p μi i 组元在相p 中化学势,则多种晶核析出时体系自由能可表示为: ...
... 尽管SFFK模型中仍存在一些可调的未知参量(例如γp 、p Di Mp 、λ p 等),其已被植入到Matcalc软件[64 ] 中,成为除TC-PRISMA(即KWN模型)外又一广泛应用的析出相模拟工具[65 ~67 ] .图6 [29 ] 为采用不同界面能得到的600℃下Fe-Mn-Si-Cr-Mo-Ti-C体系中各种Mx Cy M 2 C),因此构建可靠、准确的热-动力学数据库对于析出模拟至关重要. ...
... [
29 ]
The obtained precipitation sequences in a Fe-Mn-Si-Cr-Mo-Ti-C system at 600℃ via SFFK model using different interface energies<sup>[<xref ref-type="bibr" rid="R29">29</xref>]</sup> Fig.6 ![]()
<strong>3 </strong>多尺度热<strong>-</strong>动力学指引下的析出相研究 CALPHAD数据库中相平衡数据比热化学数据更多一些,原因在于前者更易从实验获得[31 ] .随着计算材料学发展,第一性原理计算已成为获取热力学数据的有效途径.第一性原理可与CALPHAD计算以统计热力学为桥梁相结合,一方面补充CALPHAD中缺乏的数据,另一方面提升模型的预测性能.由于相变时材料结构的多时空间尺度变化[68 ] ,多尺度组织模拟对于理解与每个尺度下相关的性能变化至关重要,从第一性原理原子尺度出发的计算便为多尺度模拟提供了可能.本节简述第一性原理计算在析出相研究中的应用,分别介绍基于第一性原理计算的多尺度相场法模拟和基于Fokker-Planck方程的多尺度模拟. ...
... [
29 ]
Fig.6 ![]()
<strong>3 </strong>多尺度热<strong>-</strong>动力学指引下的析出相研究 CALPHAD数据库中相平衡数据比热化学数据更多一些,原因在于前者更易从实验获得[31 ] .随着计算材料学发展,第一性原理计算已成为获取热力学数据的有效途径.第一性原理可与CALPHAD计算以统计热力学为桥梁相结合,一方面补充CALPHAD中缺乏的数据,另一方面提升模型的预测性能.由于相变时材料结构的多时空间尺度变化[68 ] ,多尺度组织模拟对于理解与每个尺度下相关的性能变化至关重要,从第一性原理原子尺度出发的计算便为多尺度模拟提供了可能.本节简述第一性原理计算在析出相研究中的应用,分别介绍基于第一性原理计算的多尺度相场法模拟和基于Fokker-Planck方程的多尺度模拟. ...
Multi-scale modeling of the complex microstructural evolution in structural phase transformations
7
2019
... 材料组织结构演化属于热力学非平衡过程,对此,可用动力学方程来描述体系内部状态变量,例如Fick第二定律描述了扩散场在时空间的演化.对于复杂体系,则需同时考虑置换、间隙原子与空位的运动,精确求解扩散场尤为困难.热力学极值原理是处理该复杂情形的有效工具,据其推导的演化控制方程因形式简单也非偏微分方程,既可弥补传统唯象模型的复杂性也能得到具有物理意义的解.该方法最早由Onsager[22 ] 在1931年构建,在30年后被Ziegler[23 ,24 ] 推广为处理非平衡热力学体系的变分原理,此后出现了很多其他变分形式,具体可见文献[25 ~27 ].本文介绍与SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型[28 ,29 ] 及本团队构建的多尺度组织预测模型[30 ] 相关的Ziegler热力学极值原理(亦称“最大熵产生原理”)在微观结构预测中的应用. ...
... 为解决析出反应中多尺度和多相竞争问题,本团队考虑热-动力学协同作用,结合最大熵产生原理和统计力学构建了从原子尺度计算出发,无可调参数的多尺度组织预测模型[30 ] .将介观体系视为由微观代表体积元构成的系综,体积元状态用其内部析出物的微观结构参量κi (某一维度尺寸、体积、表面积或成分)来描述.不同状态体积元在系综中的占比用概率密度f p 表示,因此f p 是析出物微观结构参量的函数,f p 的分布表征了整个介观体系的状态并唯一对应体系自由能.不同种类析出物的演化过程对应着体系不同的自由能耗散路径,那么根据最大熵产生原理,体系会自发在这些潜在路径间做即时选择,以使相变每时每刻的能量耗散速率最大.经过热力学极值原理的变分处理后,所得体系演化方程具有Fokker-Planck方程的形式: ...
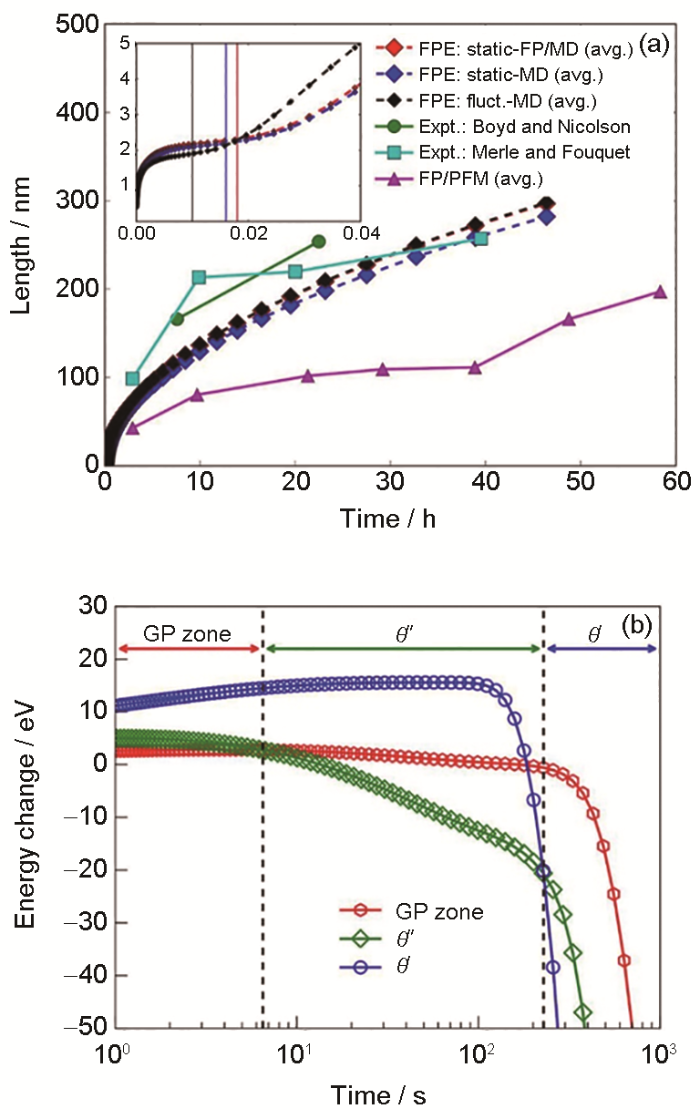
... 该模型正确而可靠地模拟了铝合金的析出过程.对于Al-2%Cu (原子分数)合金[30 ] :基于晶体学信息构建出原子构型后,采用第一性原理和分子动力学模拟得到Φ ;可通过变分过渡态理论计算速率常数获得Mij ,为简化计算,用其与扩散系数间的关系6 D i a 2 S i * S i * i 组分原子数).通过交替方向隐式有限差分法求解式(41) 得到结论:图10 a[30 ] 中该模型所得θ' 尺寸随时间的变化(红、蓝、黑色线)比相场法结果(紫色线)更加吻合实验数据(绿、浅蓝色线);图10 b[30 ] 中的能量耗散路径体现出在实验中被大量证实的GP区→θ" →θ' 析出序列.类似地,模拟得到了与实验数据吻合的Al-Mg-Si合金中β" 析出动力学[104 ] . ...
... [30 ]中该模型所得θ' 尺寸随时间的变化(红、蓝、黑色线)比相场法结果(紫色线)更加吻合实验数据(绿、浅蓝色线);图10 b[30 ] 中的能量耗散路径体现出在实验中被大量证实的GP区→θ" →θ' 析出序列.类似地,模拟得到了与实验数据吻合的Al-Mg-Si合金中β" 析出动力学[104 ] . ...
... [30 ]中的能量耗散路径体现出在实验中被大量证实的GP区→θ" →θ' 析出序列.类似地,模拟得到了与实验数据吻合的Al-Mg-Si合金中β" 析出动力学[104 ] . ...
... [
30 ]
The size evolution of <i>θ'</i> at 473 K (a) and the precipitation sequence, <i>i.e.</i> GP zone→<i>θ"</i>→<i>θ' </i>(b) in Al-2%Cu (atomic fraction) alloy (The abbreviations FP, MD, FPE, and PFM represent first-principles, molecular dynamics, Fokker-Planck equation, and phase field method, respectively)<sup>[<xref ref-type="bibr" rid="R30">30</xref>]</sup> Fig.10 ![]()
该模型随后被进一步推广应用于钢铁中,并用于探究实验中无法得知的析出机理.本团队[58 ] 通过修正Mij 计算方法(使恒定化学计量的析出相界面处原子吸附动力学更加准确),模拟了碳钢在低温回火中过渡碳化物ε -Fe2 C和η -Fe2 C的竞争析出.虽然在实验中先后发现了六方晶格的ε 和正交晶格的η ,但由于这2种晶体结构具有相同的Fe原子排布位置与不同的C原子排布方式,因而利用有限的实验技术难以区分,于是,过渡碳化物的真实结构成为了未解之谜.图11 [58 ] 所示的473 K下析出模拟结果显示出ε →η 的析出序列,表明这2种结构都会出现.图12 [58 ] 显示出不同温度下热力学驱动力、动力学能垒和析出组织间的相互关联:随着析出进行,热力学驱动力不断减小,动力学能垒不断增大;当温度提升后,析出物长大到同一尺寸时对应的热力学驱动力增大,动力学能垒减小.这说明热力学驱动力和动力学能垒以此消彼长的变化形式(即热-动力学相关性)来协同控制析出反应. ...
... [
30 ]
Fig.10 ![]()
该模型随后被进一步推广应用于钢铁中,并用于探究实验中无法得知的析出机理.本团队[58 ] 通过修正Mij 计算方法(使恒定化学计量的析出相界面处原子吸附动力学更加准确),模拟了碳钢在低温回火中过渡碳化物ε -Fe2 C和η -Fe2 C的竞争析出.虽然在实验中先后发现了六方晶格的ε 和正交晶格的η ,但由于这2种晶体结构具有相同的Fe原子排布位置与不同的C原子排布方式,因而利用有限的实验技术难以区分,于是,过渡碳化物的真实结构成为了未解之谜.图11 [58 ] 所示的473 K下析出模拟结果显示出ε →η 的析出序列,表明这2种结构都会出现.图12 [58 ] 显示出不同温度下热力学驱动力、动力学能垒和析出组织间的相互关联:随着析出进行,热力学驱动力不断减小,动力学能垒不断增大;当温度提升后,析出物长大到同一尺寸时对应的热力学驱动力增大,动力学能垒减小.这说明热力学驱动力和动力学能垒以此消彼长的变化形式(即热-动力学相关性)来协同控制析出反应. ...
First-principles calculations and CALPHAD modeling of thermodynamics
4
2009
... 由于CALPHAD计算[31 ,32 ] 在预测微观组织形成中的重要作用,集成计算材料工程与材料基因组计划提出发展CALPHAD技术与构建高质量CALPHAD数据库的迫切需求[33 ,34 ] .继Andersson和Ågren[35 ] 提出多组分扩散问题的数值求解后,基于CALPHAD的扩散相变模拟便被大量开展,CALPHAD发展历程可见文献[36 ].本节介绍基于CALPHAD数据库的介观尺度析出模拟,包括DIffusion-Controlled TRAnsformation (DICTRA)软件、基于形核长大粗化理论的模拟(例如KWN (Kampmann-Wagner numerical)模型)、基于热力学极值原理的SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型. ...
... CALPHAD数据库中相平衡数据比热化学数据更多一些,原因在于前者更易从实验获得[31 ] .随着计算材料学发展,第一性原理计算已成为获取热力学数据的有效途径.第一性原理可与CALPHAD计算以统计热力学为桥梁相结合,一方面补充CALPHAD中缺乏的数据,另一方面提升模型的预测性能.由于相变时材料结构的多时空间尺度变化[68 ] ,多尺度组织模拟对于理解与每个尺度下相关的性能变化至关重要,从第一性原理原子尺度出发的计算便为多尺度模拟提供了可能.本节简述第一性原理计算在析出相研究中的应用,分别介绍基于第一性原理计算的多尺度相场法模拟和基于Fokker-Planck方程的多尺度模拟. ...
... 然而,以上应用都基于0 K,实际析出发生在有限温度下.有些析出物的基态不稳定,但可能因有限温度下的声子和电子激发变得稳定,因此有限温度下的热力学性能获取很有必要.有限温度下的自由能计算需考虑以下部分的贡献:0 K下的内能;原子尺度下各组分混合的构型熵;晶格振动对熵的贡献;电子热激发和自旋极化对熵的贡献[31 ] .对于理想混合,可直接由(-R Σxi lnxi )得到构型熵(xi 为i 组分摩尔分数),若存在短程有序,可用团簇扩张(cluster expansion)和Monte Carlo模拟[79 ,80 ] 来处理.通过简谐或准简谐近似可从DFT计算中获得声子谱,继而得到振动熵[31 ] .Wang等[81 ,82 ] 通过考虑有限温度下不同磁性状态的混合得到电子尺度的熵贡献.同样以碳化物为例.Fang等[83 ] 计算了Fe3 C和Fe2 C间相对自由能差随温度的变化,结果显示,当温度升高到330 K,稳定结构从Fe2 C变为Fe3 C,这与实验结果吻合,并说明Fe3 C的普遍存在由晶格振动和磁序变化所致.Kaplan等[84 ] 计算了Cr7 C3 的2种晶格结构的相对稳定性随温度的变化,表明低温条件下Cr7 C3 是六方结构(P 63 mc ),当温度升到470 K,会向正交结构(Pnma )转变.从以上热力学角度只可做初步分析,界面能和应变能会对析出过程产生很大影响,因此,考虑热-动力学协同的模拟会更有价值. ...
... [31 ].Wang等[81 ,82 ] 通过考虑有限温度下不同磁性状态的混合得到电子尺度的熵贡献.同样以碳化物为例.Fang等[83 ] 计算了Fe3 C和Fe2 C间相对自由能差随温度的变化,结果显示,当温度升高到330 K,稳定结构从Fe2 C变为Fe3 C,这与实验结果吻合,并说明Fe3 C的普遍存在由晶格振动和磁序变化所致.Kaplan等[84 ] 计算了Cr7 C3 的2种晶格结构的相对稳定性随温度的变化,表明低温条件下Cr7 C3 是六方结构(P 63 mc ),当温度升到470 K,会向正交结构(Pnma )转变.从以上热力学角度只可做初步分析,界面能和应变能会对析出过程产生很大影响,因此,考虑热-动力学协同的模拟会更有价值. ...
1
1998
... 由于CALPHAD计算[31 ,32 ] 在预测微观组织形成中的重要作用,集成计算材料工程与材料基因组计划提出发展CALPHAD技术与构建高质量CALPHAD数据库的迫切需求[33 ,34 ] .继Andersson和Ågren[35 ] 提出多组分扩散问题的数值求解后,基于CALPHAD的扩散相变模拟便被大量开展,CALPHAD发展历程可见文献[36 ].本节介绍基于CALPHAD数据库的介观尺度析出模拟,包括DIffusion-Controlled TRAnsformation (DICTRA)软件、基于形核长大粗化理论的模拟(例如KWN (Kampmann-Wagner numerical)模型)、基于热力学极值原理的SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型. ...
CALPHAD, first and second generation—Birth of the materials genome
1
2014
... 由于CALPHAD计算[31 ,32 ] 在预测微观组织形成中的重要作用,集成计算材料工程与材料基因组计划提出发展CALPHAD技术与构建高质量CALPHAD数据库的迫切需求[33 ,34 ] .继Andersson和Ågren[35 ] 提出多组分扩散问题的数值求解后,基于CALPHAD的扩散相变模拟便被大量开展,CALPHAD发展历程可见文献[36 ].本节介绍基于CALPHAD数据库的介观尺度析出模拟,包括DIffusion-Controlled TRAnsformation (DICTRA)软件、基于形核长大粗化理论的模拟(例如KWN (Kampmann-Wagner numerical)模型)、基于热力学极值原理的SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型. ...
Materials genomics: From CALPHAD to flight
1
2014
... 由于CALPHAD计算[31 ,32 ] 在预测微观组织形成中的重要作用,集成计算材料工程与材料基因组计划提出发展CALPHAD技术与构建高质量CALPHAD数据库的迫切需求[33 ,34 ] .继Andersson和Ågren[35 ] 提出多组分扩散问题的数值求解后,基于CALPHAD的扩散相变模拟便被大量开展,CALPHAD发展历程可见文献[36 ].本节介绍基于CALPHAD数据库的介观尺度析出模拟,包括DIffusion-Controlled TRAnsformation (DICTRA)软件、基于形核长大粗化理论的模拟(例如KWN (Kampmann-Wagner numerical)模型)、基于热力学极值原理的SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型. ...
Models for numerical treatment of multicomponent diffusion in simple phases
2
1992
... 由于CALPHAD计算[31 ,32 ] 在预测微观组织形成中的重要作用,集成计算材料工程与材料基因组计划提出发展CALPHAD技术与构建高质量CALPHAD数据库的迫切需求[33 ,34 ] .继Andersson和Ågren[35 ] 提出多组分扩散问题的数值求解后,基于CALPHAD的扩散相变模拟便被大量开展,CALPHAD发展历程可见文献[36 ].本节介绍基于CALPHAD数据库的介观尺度析出模拟,包括DIffusion-Controlled TRAnsformation (DICTRA)软件、基于形核长大粗化理论的模拟(例如KWN (Kampmann-Wagner numerical)模型)、基于热力学极值原理的SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型. ...
... 式中,Dkj 为k 组分和j 组分间的互扩散系数(interdiffusivity),Dkn 为k 组分和n 组分间的互扩散系数,D ˜ k j n Vj 和Vn 分别为第j 个和第n 个组分的偏摩尔体积.DICTRA模拟取决于热-动力学数据库的质量(例如扩散系数的准确度).Andersson和Ågren[35 ] 基于体积固定参考框架给出: ...
A brief history of CALPHAD
1
2008
... 由于CALPHAD计算[31 ,32 ] 在预测微观组织形成中的重要作用,集成计算材料工程与材料基因组计划提出发展CALPHAD技术与构建高质量CALPHAD数据库的迫切需求[33 ,34 ] .继Andersson和Ågren[35 ] 提出多组分扩散问题的数值求解后,基于CALPHAD的扩散相变模拟便被大量开展,CALPHAD发展历程可见文献[36 ].本节介绍基于CALPHAD数据库的介观尺度析出模拟,包括DIffusion-Controlled TRAnsformation (DICTRA)软件、基于形核长大粗化理论的模拟(例如KWN (Kampmann-Wagner numerical)模型)、基于热力学极值原理的SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型. ...
CALPHAD—Type modeling of diffusion kinetics in multicomponent alloys
3
2017
... Thermal-Calc Software AB公司发行的DICTRA软件可基于局域平衡和尖锐界面假设对多组分扩散方程求数值解,以完成复杂合金的扩散析出模拟.虽然DICTRA可处理复杂体系中扩散过程,但只适用于一维情形,即可简化成一维的几何形状(无限大且具有一定厚度的平板、无限长且具有一定大小底面的圆柱和具有一定半径的球体),模拟边界条件可根据问题灵活选择.DICTRA包含以下模块:(i) 扩散方程求解;(ii) 热力学平衡计算;(iii) 流守恒方程求解;(iv) 相界面位置移动和网格划分调整.如图2 [37 ] 所示,主要工作为求解多组分扩散方程[22 ] : ...
... [
37 ]
Work flow for DICTRA simulation<sup>[<xref ref-type="bibr" rid="R37">37</xref>]</sup> Fig.2 ![]()
∂ c k ∂ t = ∇ ∑ j = 1 n - 1 D ˜ k j n ∇ c j (20) D ˜ k j n = D k j - D k n V j V n (21) 式中,Dkj 为k 组分和j 组分间的互扩散系数(interdiffusivity),Dkn 为k 组分和n 组分间的互扩散系数,D ˜ k j n Vj 和Vn 分别为第j 个和第n 个组分的偏摩尔体积.DICTRA模拟取决于热-动力学数据库的质量(例如扩散系数的准确度).Andersson和Ågren[35 ] 基于体积固定参考框架给出: ...
... [
37 ]
Fig.2 ![]()
∂ c k ∂ t = ∇ ∑ j = 1 n - 1 D ˜ k j n ∇ c j (20) D ˜ k j n = D k j - D k n V j V n (21) 式中,Dkj 为k 组分和j 组分间的互扩散系数(interdiffusivity),Dkn 为k 组分和n 组分间的互扩散系数,D ˜ k j n Vj 和Vn 分别为第j 个和第n 个组分的偏摩尔体积.DICTRA模拟取决于热-动力学数据库的质量(例如扩散系数的准确度).Andersson和Ågren[35 ] 基于体积固定参考框架给出: ...
Characterization of carbides in steels using atom probe field-ion microscopy
1
2000
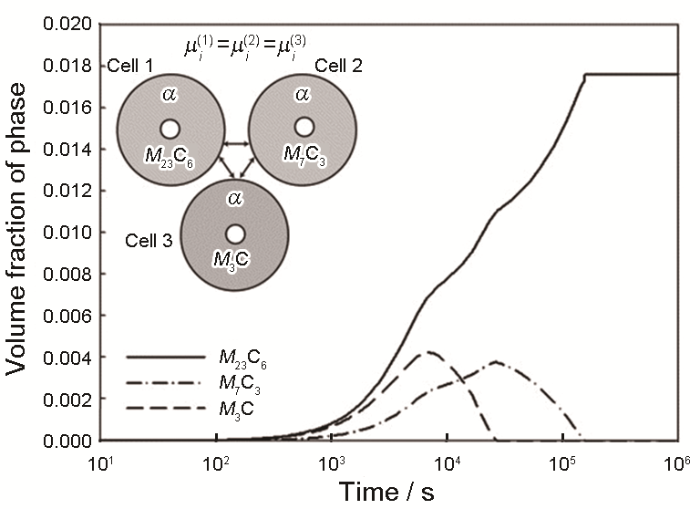
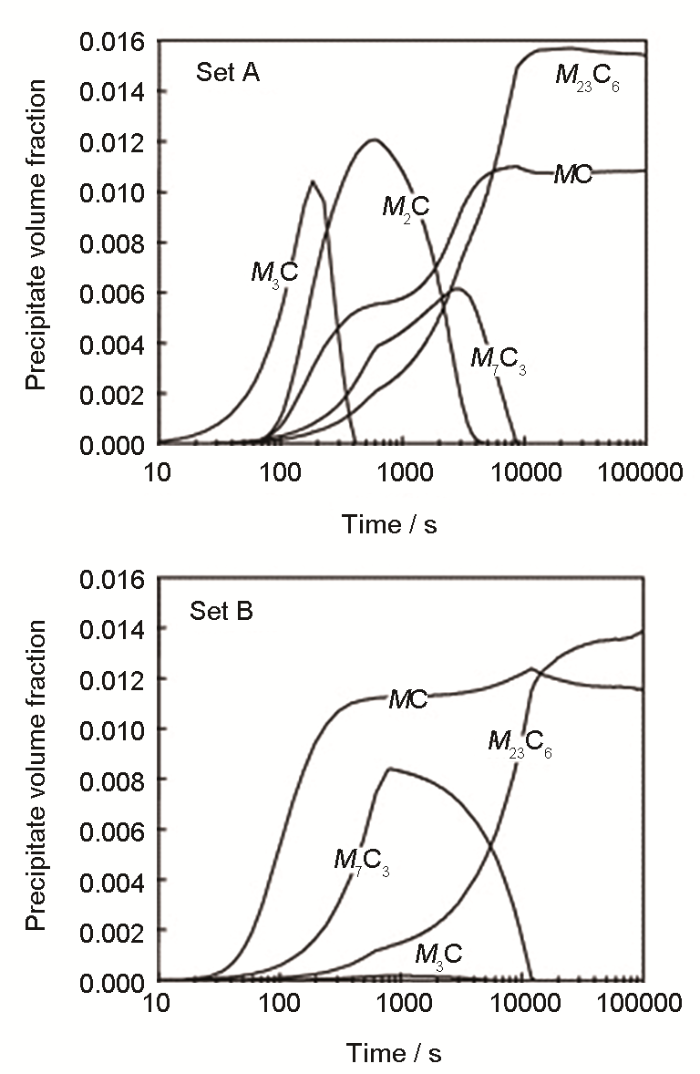
... 除Thermo-Calc外,其他热化学软件,例如FactSage、CEQCSI、MPE、MTDATA,均涉及铁基、铝基、镁基、镍基、钛基、铜基、硅基、锆基合金的计算.动力学Mi 数据库发展相对缓慢,但对于铁基、铝基的金属材料已较为完善.DICTRA的应用价值可以通过钢铁中碳化物析出模拟结果得以证实.含铬钢在高温回火时会析出M 3 C、M 7 C3 、M 23 C6 等[38 ] ,如图3 [39 ] 所示,Schneider和Inden[39 ] 用DICTRA模拟了1053 K下Fe-12Cr-0.1C合金中这些碳化物的竞争析出,与实验结果一致,由于M 3 C和M 7 C3 溶解,得到了稳定的M 23 C6 ;图中3个cell分别包含了一种碳化物,基体中化学势相等维持了cell间的联系.该模拟虽进行了全扩散场的求解,但无法考虑形核,且cell不足以代表整个介观体系,无法得到尺寸分布等介观参量. ...
Simulation of the kinetics of precipitation reactions in ferritic steels
4
2005
... 除Thermo-Calc外,其他热化学软件,例如FactSage、CEQCSI、MPE、MTDATA,均涉及铁基、铝基、镁基、镍基、钛基、铜基、硅基、锆基合金的计算.动力学Mi 数据库发展相对缓慢,但对于铁基、铝基的金属材料已较为完善.DICTRA的应用价值可以通过钢铁中碳化物析出模拟结果得以证实.含铬钢在高温回火时会析出M 3 C、M 7 C3 、M 23 C6 等[38 ] ,如图3 [39 ] 所示,Schneider和Inden[39 ] 用DICTRA模拟了1053 K下Fe-12Cr-0.1C合金中这些碳化物的竞争析出,与实验结果一致,由于M 3 C和M 7 C3 溶解,得到了稳定的M 23 C6 ;图中3个cell分别包含了一种碳化物,基体中化学势相等维持了cell间的联系.该模拟虽进行了全扩散场的求解,但无法考虑形核,且cell不足以代表整个介观体系,无法得到尺寸分布等介观参量. ...
... [39 ]用DICTRA模拟了1053 K下Fe-12Cr-0.1C合金中这些碳化物的竞争析出,与实验结果一致,由于M 3 C和M 7 C3 溶解,得到了稳定的M 23 C6 ;图中3个cell分别包含了一种碳化物,基体中化学势相等维持了cell间的联系.该模拟虽进行了全扩散场的求解,但无法考虑形核,且cell不足以代表整个介观体系,无法得到尺寸分布等介观参量. ...
... [
39 ]
Three-cell simulation of competitive growth of stable <i>M</i><sub>23</sub>C<sub>6</sub> and of metastable <i>M</i><sub>7</sub>C<sub>3</sub> and <i>M</i><sub>3</sub>C (<i>M</i> represents the substitutional alloying elements) in Fe-12Cr-0.1C at 1053 K (<i>μ<sub>i</sub></i>—chemical potential of component <i>i</i>)<sup>[<xref ref-type="bibr" rid="R39">39</xref>]</sup> Fig.3 ![]()
DICTRA也可用于粗化研究[40 ~43 ] ,假设析出物尺寸服从粗化阶段的LSW分布,即最大微粒尺寸是平均微粒尺寸的1.5倍,通过模拟具有体系中最大尺寸的微粒的长大便可估计整个体系的粗化,同时可得到平均微粒尺寸.由于尚缺乏完整的析出物界面能数据,粗化模拟时常将其作为可调参数,对结果造成很大影响. ...
... [
39 ]
Fig.3 ![]()
DICTRA也可用于粗化研究[40 ~43 ] ,假设析出物尺寸服从粗化阶段的LSW分布,即最大微粒尺寸是平均微粒尺寸的1.5倍,通过模拟具有体系中最大尺寸的微粒的长大便可估计整个体系的粗化,同时可得到平均微粒尺寸.由于尚缺乏完整的析出物界面能数据,粗化模拟时常将其作为可调参数,对结果造成很大影响. ...
Simulations of coarsening behavior for M 23 C6 carbides in AISI H13 steel
1
2006
... DICTRA也可用于粗化研究[40 ~43 ] ,假设析出物尺寸服从粗化阶段的LSW分布,即最大微粒尺寸是平均微粒尺寸的1.5倍,通过模拟具有体系中最大尺寸的微粒的长大便可估计整个体系的粗化,同时可得到平均微粒尺寸.由于尚缺乏完整的析出物界面能数据,粗化模拟时常将其作为可调参数,对结果造成很大影响. ...
Complex carbide growth, dissolution, and coarsening in a modified 12 pct chromium steel—An experimental and theoretical study
2001
Diffusion simulations of M C and M 7 C3 carbide coarsening in bcc and fcc matrix utilising new thermodynamic and kinetic description
2008
Precipitation kinetics in a 10.5%Cr heat resistant steel: Experimental results and simulation by TC-PRISMA/DICTRA
1
2017
... DICTRA也可用于粗化研究[40 ~43 ] ,假设析出物尺寸服从粗化阶段的LSW分布,即最大微粒尺寸是平均微粒尺寸的1.5倍,通过模拟具有体系中最大尺寸的微粒的长大便可估计整个体系的粗化,同时可得到平均微粒尺寸.由于尚缺乏完整的析出物界面能数据,粗化模拟时常将其作为可调参数,对结果造成很大影响. ...
Precipitation and strengthening modeling for disk-shaped particles in aluminum alloys: Size distribution considered
2
2018
... 微观结构模拟方法可划分为直接精细数值(direct detailed numerical)法和频率分布函数(frequency distribution function)法[44 ] .前者以相场法为代表;后者通常指基于经典形核-长大-粗化理论的模拟,通过对结构特征的统计处理来完整地描述析出全过程.在频率分布函数法中,可依据形核长大速率方程的应用方式进一步划分为平均尺寸法与尺寸分级法[11 ] ,此为本节重点. ...
... 式中,C 0 为初始基体溶质浓度.该方法开创了尺寸分级法的先河,通过将图4 的微粒尺寸分布f (R )离散为许多部分,即划分尺寸等级,然后将尺寸的连续时间演化离散为单个时间步进行.KWN模型随后被不断改进,Myhr和Grong[49 ] 提出了计算相邻尺寸等级之间通量的有限差分方法,如图5 [11 ] 所示,也称作“类Euler法”.Nicolas和Deschamps[50 ] 将KWN模型应用到非等温过程中.Du等[51 ] 改善了长大速率方程,考虑高过饱和度情形和交叉扩散后,通过求解界面移动速率、变化的tie-line和物质流守恒方程使得KWN模型可得到多组分析出物的成分变化.由于实际晶核往往是非球形的[52 ~54 ] ,不同形状晶核因伴随不同的应变能和界面能变化而具有不同的析出过程,模拟需基于晶核的真实形貌.于是Holmedal等[55 ] 和Li等[44 ] 通过引入长大速率和Gibbs-Thomson效应的修正因子将KWN模型用于处理非球形析出相. ...
Kinetics of nucleation in near-critical fluids
1
1980
... 平均尺寸法中最典型的代表是,Langer和Schwartz[45 ] 在1980年提出的将形核、长大、粗化在同一框架下处理的LS模型.他们假定体系单位体积内含有N LS 个平均尺寸为R ¯ L S f (R )演化服从: ...
Modelling the age-hardening precipitation by a revised Langer and Schwartz approach with log-normal size distribution
4
2020
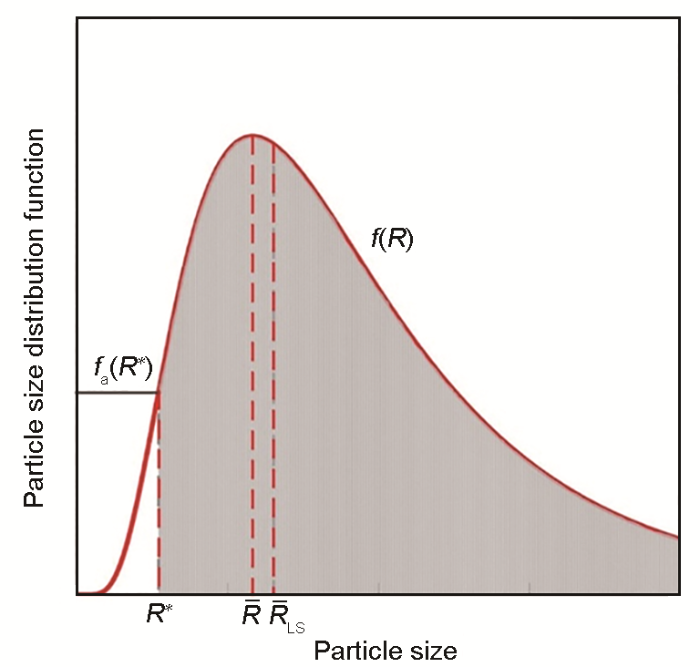
... 式中,b 为常数.如图4 [46 ] 所示,在LS理论中只有半径超过R * 的微粒才被算作新相,于是R ¯ L S
... [
46 ]
Log-normal particle size distribution with a mean particle radius <span class="formulaText"><inline-formula><math id="M48"><mover accent="true"><mrow><mi>R</mi></mrow><mo stretchy="true">¯</mo></mover></math></span></inline-formula></span> (Particles with radius <i>R</i><<i>R</i><sup>*</sup> are continuously dissolving in the matrix. In the Langer and Schwartz approach, only the hatched area with particle radius <i>R</i>><i>R</i><sup>*</sup> contributes to the mean particle radius <span class="formulaText"><inline-formula><math id="M49"><msub><mrow><mover accent="true"><mrow><mi>R</mi></mrow><mo stretchy="true">¯</mo></mover></mrow><mrow><mi mathvariant="normal">L</mi><mi mathvariant="normal">S</mi></mrow></msub></math></span></inline-formula></span><span class="formulaNumber">.</span> <span class="formulaText"><inline-formula><math id="M50"><mover accent="true"><mrow><mi>R</mi></mrow><mo stretchy="true">¯</mo></mover></math></span></inline-formula></span> is the mean radius for the full log-normal distribution, <span class="formulaText"><inline-formula><math id="M51"><msub><mrow><mover accent="true"><mrow><mi>R</mi></mrow><mo stretchy="true">¯</mo></mover></mrow><mrow><mi mathvariant="normal">L</mi><mi mathvariant="normal">S</mi></mrow></msub></math></span></inline-formula></span> is the mean radius of the stable particles (<i>R</i>><i>R</i><sup>*</sup>), <i>f</i>(<i>R</i>) is the particle size distribution and <i>f</i><sub>a</sub>(<i>R<sup>*</sup></i>) is the density of the particles with the critical nucleation size)<sup>[<xref ref-type="bibr" rid="R46">46</xref>]</sup> Fig.4 ![]()
R ¯ L S = 1 N L S ∫ R * ∞ f R R d R (26) d R ¯ L S d t = v R ¯ L S + R ¯ L S - R * f a R * N L S d R * d t + R * + δ R * - R ¯ L S J R * N L S (27) 式中,δR * 表示新形成的析出物尺寸稍大于临界尺寸的部分,式(27) 右边第1项由式(7) 求得,第2和3项分别表示微粒溶解和形核引起的平均尺寸变化.由形核和溶解引起的析出物数量变化d N L S d t
... [
46 ]
Fig.4 ![]()
R ¯ L S = 1 N L S ∫ R * ∞ f R R d R (26) d R ¯ L S d t = v R ¯ L S + R ¯ L S - R * f a R * N L S d R * d t + R * + δ R * - R ¯ L S J R * N L S (27) 式中,δR * 表示新形成的析出物尺寸稍大于临界尺寸的部分,式(27) 右边第1项由式(7) 求得,第2和3项分别表示微粒溶解和形核引起的平均尺寸变化.由形核和溶解引起的析出物数量变化d N L S d t
... 式中,c ¯ 25 )、(27 )~(29 )联立即可描述平均尺寸及数量演化.然而,LS理论中求解界面成分时采用线性形式的Gibbs-Thomson方程,不适用于小微粒,后来Kampmann和Wagner通过引入非线性Gibbs-Thomson方程(式(8) )修正了LS模型,即著名的MLS (modified Langer-Schwartz)理论[9 ,46 ,47 ] .平均尺寸法虽计算量小,但无法描述微粒尺寸分布[48 ] ,而该参量可借助尺寸分级法获得. ...
Precipitation kinetics in metastable solid solutions—Theoretical considerations and application to Cu-Ti alloys
1
1987
... 式中,c ¯ 25 )、(27 )~(29 )联立即可描述平均尺寸及数量演化.然而,LS理论中求解界面成分时采用线性形式的Gibbs-Thomson方程,不适用于小微粒,后来Kampmann和Wagner通过引入非线性Gibbs-Thomson方程(式(8) )修正了LS模型,即著名的MLS (modified Langer-Schwartz)理论[9 ,46 ,47 ] .平均尺寸法虽计算量小,但无法描述微粒尺寸分布[48 ] ,而该参量可借助尺寸分级法获得. ...
Modelling the evolution of particle size distribution during nucleation, growth and coarsening
2
2004
... 式中,c ¯ 25 )、(27 )~(29 )联立即可描述平均尺寸及数量演化.然而,LS理论中求解界面成分时采用线性形式的Gibbs-Thomson方程,不适用于小微粒,后来Kampmann和Wagner通过引入非线性Gibbs-Thomson方程(式(8) )修正了LS模型,即著名的MLS (modified Langer-Schwartz)理论[9 ,46 ,47 ] .平均尺寸法虽计算量小,但无法描述微粒尺寸分布[48 ] ,而该参量可借助尺寸分级法获得. ...
... KWN模型已成为Thermal-Calc软件中TC-PRISMA模块的核心理论,被广泛应用于复杂工业合金的析出相模拟、析出机理探究与经典理论发展.微粒尺寸分布,即一定尺寸范围内的析出物数量,对金属性能有很大影响.Robson[48 ] 借助KWN模型模拟了Cu-1.02%Co (原子分数)合金600℃下的析出反应,结果表明早期存在着晶核尺寸的双峰分布,是由形核与长大速率变化引起的,该现象也在其他实验与模拟中发现[49 ,56 ~58 ] .Robson[59 ] 还研究了形核、长大、粗化阶段出现的重叠现象,结果表明重叠程度与界面能、溶质过饱和度有很大关联而与溶质扩散性无关. ...
Modelling of non-isothermal transformations in alloys containing a particle distribution
2
2000
... 式中,C 0 为初始基体溶质浓度.该方法开创了尺寸分级法的先河,通过将图4 的微粒尺寸分布f (R )离散为许多部分,即划分尺寸等级,然后将尺寸的连续时间演化离散为单个时间步进行.KWN模型随后被不断改进,Myhr和Grong[49 ] 提出了计算相邻尺寸等级之间通量的有限差分方法,如图5 [11 ] 所示,也称作“类Euler法”.Nicolas和Deschamps[50 ] 将KWN模型应用到非等温过程中.Du等[51 ] 改善了长大速率方程,考虑高过饱和度情形和交叉扩散后,通过求解界面移动速率、变化的tie-line和物质流守恒方程使得KWN模型可得到多组分析出物的成分变化.由于实际晶核往往是非球形的[52 ~54 ] ,不同形状晶核因伴随不同的应变能和界面能变化而具有不同的析出过程,模拟需基于晶核的真实形貌.于是Holmedal等[55 ] 和Li等[44 ] 通过引入长大速率和Gibbs-Thomson效应的修正因子将KWN模型用于处理非球形析出相. ...
... KWN模型已成为Thermal-Calc软件中TC-PRISMA模块的核心理论,被广泛应用于复杂工业合金的析出相模拟、析出机理探究与经典理论发展.微粒尺寸分布,即一定尺寸范围内的析出物数量,对金属性能有很大影响.Robson[48 ] 借助KWN模型模拟了Cu-1.02%Co (原子分数)合金600℃下的析出反应,结果表明早期存在着晶核尺寸的双峰分布,是由形核与长大速率变化引起的,该现象也在其他实验与模拟中发现[49 ,56 ~58 ] .Robson[59 ] 还研究了形核、长大、粗化阶段出现的重叠现象,结果表明重叠程度与界面能、溶质过饱和度有很大关联而与溶质扩散性无关. ...
Characterisation and modelling of precipitate evolution in an Al-Zn-Mg alloy during non-isothermal heat treatments
1
2003
... 式中,C 0 为初始基体溶质浓度.该方法开创了尺寸分级法的先河,通过将图4 的微粒尺寸分布f (R )离散为许多部分,即划分尺寸等级,然后将尺寸的连续时间演化离散为单个时间步进行.KWN模型随后被不断改进,Myhr和Grong[49 ] 提出了计算相邻尺寸等级之间通量的有限差分方法,如图5 [11 ] 所示,也称作“类Euler法”.Nicolas和Deschamps[50 ] 将KWN模型应用到非等温过程中.Du等[51 ] 改善了长大速率方程,考虑高过饱和度情形和交叉扩散后,通过求解界面移动速率、变化的tie-line和物质流守恒方程使得KWN模型可得到多组分析出物的成分变化.由于实际晶核往往是非球形的[52 ~54 ] ,不同形状晶核因伴随不同的应变能和界面能变化而具有不同的析出过程,模拟需基于晶核的真实形貌.于是Holmedal等[55 ] 和Li等[44 ] 通过引入长大速率和Gibbs-Thomson效应的修正因子将KWN模型用于处理非球形析出相. ...
A mathematical model coupled to CALPHAD to predict precipitation kinetics for multicomponent aluminum alloys
2
2012
... 式中,C 0 为初始基体溶质浓度.该方法开创了尺寸分级法的先河,通过将图4 的微粒尺寸分布f (R )离散为许多部分,即划分尺寸等级,然后将尺寸的连续时间演化离散为单个时间步进行.KWN模型随后被不断改进,Myhr和Grong[49 ] 提出了计算相邻尺寸等级之间通量的有限差分方法,如图5 [11 ] 所示,也称作“类Euler法”.Nicolas和Deschamps[50 ] 将KWN模型应用到非等温过程中.Du等[51 ] 改善了长大速率方程,考虑高过饱和度情形和交叉扩散后,通过求解界面移动速率、变化的tie-line和物质流守恒方程使得KWN模型可得到多组分析出物的成分变化.由于实际晶核往往是非球形的[52 ~54 ] ,不同形状晶核因伴随不同的应变能和界面能变化而具有不同的析出过程,模拟需基于晶核的真实形貌.于是Holmedal等[55 ] 和Li等[44 ] 通过引入长大速率和Gibbs-Thomson效应的修正因子将KWN模型用于处理非球形析出相. ...
... (2) 扩散场胞法.前述模型虽被广泛应用于不同情景,但均未考虑元素在基体中的扩散交互作用及不同析出物扩散场的重叠作用.这2种效应对析出动力学均会造成影响,Du等[51 ] 采用热力学极值原理构建出析出物演化模型,证实了考虑软碰撞的重要性.本小节介绍Popov等[60 ~62 ] 提出的同时考虑这2种效应的新模型(此处称作“扩散场胞法”). ...
Composition of β ″ precipitates in Al-Mg-Si alloys by atom probe tomography and first principles calculations
1
2009
... 式中,C 0 为初始基体溶质浓度.该方法开创了尺寸分级法的先河,通过将图4 的微粒尺寸分布f (R )离散为许多部分,即划分尺寸等级,然后将尺寸的连续时间演化离散为单个时间步进行.KWN模型随后被不断改进,Myhr和Grong[49 ] 提出了计算相邻尺寸等级之间通量的有限差分方法,如图5 [11 ] 所示,也称作“类Euler法”.Nicolas和Deschamps[50 ] 将KWN模型应用到非等温过程中.Du等[51 ] 改善了长大速率方程,考虑高过饱和度情形和交叉扩散后,通过求解界面移动速率、变化的tie-line和物质流守恒方程使得KWN模型可得到多组分析出物的成分变化.由于实际晶核往往是非球形的[52 ~54 ] ,不同形状晶核因伴随不同的应变能和界面能变化而具有不同的析出过程,模拟需基于晶核的真实形貌.于是Holmedal等[55 ] 和Li等[44 ] 通过引入长大速率和Gibbs-Thomson效应的修正因子将KWN模型用于处理非球形析出相. ...
Precipitates in Al-Cu alloys revisited: Atom-probe tomographic experiments and first-principles calculations of compositional evolution and interfacial segregation
2011
Epsilon carbide precipitation during tempering of plain carbon martensite
1
1992
... 式中,C 0 为初始基体溶质浓度.该方法开创了尺寸分级法的先河,通过将图4 的微粒尺寸分布f (R )离散为许多部分,即划分尺寸等级,然后将尺寸的连续时间演化离散为单个时间步进行.KWN模型随后被不断改进,Myhr和Grong[49 ] 提出了计算相邻尺寸等级之间通量的有限差分方法,如图5 [11 ] 所示,也称作“类Euler法”.Nicolas和Deschamps[50 ] 将KWN模型应用到非等温过程中.Du等[51 ] 改善了长大速率方程,考虑高过饱和度情形和交叉扩散后,通过求解界面移动速率、变化的tie-line和物质流守恒方程使得KWN模型可得到多组分析出物的成分变化.由于实际晶核往往是非球形的[52 ~54 ] ,不同形状晶核因伴随不同的应变能和界面能变化而具有不同的析出过程,模拟需基于晶核的真实形貌.于是Holmedal等[55 ] 和Li等[44 ] 通过引入长大速率和Gibbs-Thomson效应的修正因子将KWN模型用于处理非球形析出相. ...
Precipitation of non-spherical particles in aluminum alloys part I: Generalization of the Kampmann-Wagner numerical model
1
2016
... 式中,C 0 为初始基体溶质浓度.该方法开创了尺寸分级法的先河,通过将图4 的微粒尺寸分布f (R )离散为许多部分,即划分尺寸等级,然后将尺寸的连续时间演化离散为单个时间步进行.KWN模型随后被不断改进,Myhr和Grong[49 ] 提出了计算相邻尺寸等级之间通量的有限差分方法,如图5 [11 ] 所示,也称作“类Euler法”.Nicolas和Deschamps[50 ] 将KWN模型应用到非等温过程中.Du等[51 ] 改善了长大速率方程,考虑高过饱和度情形和交叉扩散后,通过求解界面移动速率、变化的tie-line和物质流守恒方程使得KWN模型可得到多组分析出物的成分变化.由于实际晶核往往是非球形的[52 ~54 ] ,不同形状晶核因伴随不同的应变能和界面能变化而具有不同的析出过程,模拟需基于晶核的真实形貌.于是Holmedal等[55 ] 和Li等[44 ] 通过引入长大速率和Gibbs-Thomson效应的修正因子将KWN模型用于处理非球形析出相. ...
Modeling precipitation kinetics during heat treatment with calphad-based tools
1
2014
... KWN模型已成为Thermal-Calc软件中TC-PRISMA模块的核心理论,被广泛应用于复杂工业合金的析出相模拟、析出机理探究与经典理论发展.微粒尺寸分布,即一定尺寸范围内的析出物数量,对金属性能有很大影响.Robson[48 ] 借助KWN模型模拟了Cu-1.02%Co (原子分数)合金600℃下的析出反应,结果表明早期存在着晶核尺寸的双峰分布,是由形核与长大速率变化引起的,该现象也在其他实验与模拟中发现[49 ,56 ~58 ] .Robson[59 ] 还研究了形核、长大、粗化阶段出现的重叠现象,结果表明重叠程度与界面能、溶质过饱和度有很大关联而与溶质扩散性无关. ...
Influence of composition on monomodal versus multimodal γ ′ precipitation in Ni-Al-Cr alloys
2013
Modeling competitive precipitations among iron carbides during low-temperature tempering of martensitic carbon steel
9
2020
... KWN模型已成为Thermal-Calc软件中TC-PRISMA模块的核心理论,被广泛应用于复杂工业合金的析出相模拟、析出机理探究与经典理论发展.微粒尺寸分布,即一定尺寸范围内的析出物数量,对金属性能有很大影响.Robson[48 ] 借助KWN模型模拟了Cu-1.02%Co (原子分数)合金600℃下的析出反应,结果表明早期存在着晶核尺寸的双峰分布,是由形核与长大速率变化引起的,该现象也在其他实验与模拟中发现[49 ,56 ~58 ] .Robson[59 ] 还研究了形核、长大、粗化阶段出现的重叠现象,结果表明重叠程度与界面能、溶质过饱和度有很大关联而与溶质扩散性无关. ...
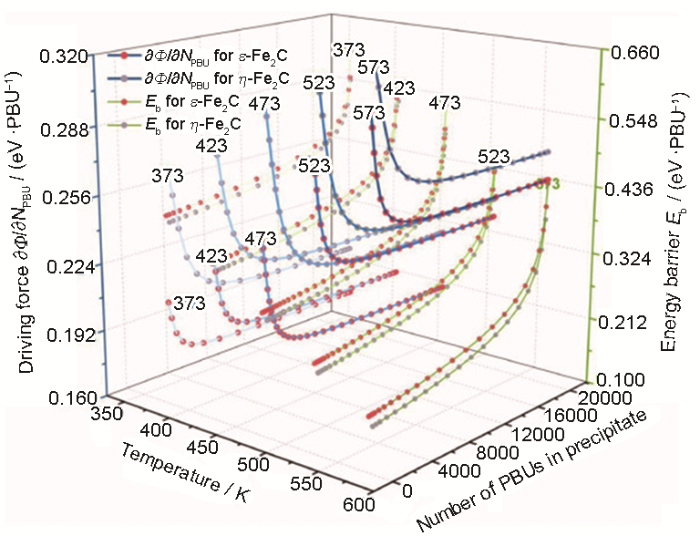
... DFT在析出热-动力学研究中发挥了重要作用,可以钢铁回火析出相(Fe2 C、M 3 C、M 7 C3 、M 23 C6 等)为例.通常,以析出物在0 K下的形成焓为热力学稳定性判据,可推断析出演化序列.基于此,Fang等[73 ] 通过比较回火过渡碳化物ε 和η 的形成焓推断出ε →η 的析出序列,该现象被相关实验[74 ] 和模拟[58 ] 所证实.形成焓也可反映成分对析出物形成的影响.Medvedeva等[75 ] 的形成焓计算表明,W和Mo的添加会使M 23 C6 更加稳定并减少其与基体间的晶格错配而有利于析出.Konyaeva等[76 ] 计算了(Fe, Cr)3 C和(Fe, Cr)7 C3 在不同置换原子浓度下的形成焓,判断出杂质原子在晶格上的优先占据位置.Ande等[77 ] 计算了合金元素在Fe3 C和铁素体间的配分焓,总结出合金元素对析出物成分变化的影响规律.利用DFT计算析出物力学性质可判断其机械稳定性.Liu等[78 ] 用应力应变法和Voigt-Reuss-Hill近似法,分别得到M 23 C6 的弹性常数和模量,表明有Fe或Mo掺杂的M 23 C6 比纯Cr23 C6 和Mn23 C6 的力学性能更好. ...
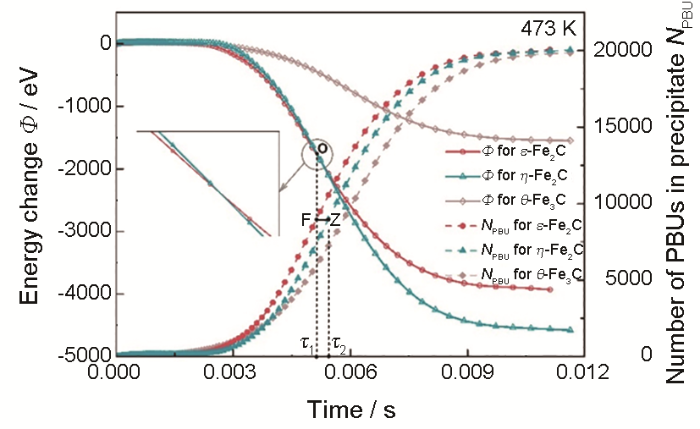
... 该模型随后被进一步推广应用于钢铁中,并用于探究实验中无法得知的析出机理.本团队[58 ] 通过修正Mij 计算方法(使恒定化学计量的析出相界面处原子吸附动力学更加准确),模拟了碳钢在低温回火中过渡碳化物ε -Fe2 C和η -Fe2 C的竞争析出.虽然在实验中先后发现了六方晶格的ε 和正交晶格的η ,但由于这2种晶体结构具有相同的Fe原子排布位置与不同的C原子排布方式,因而利用有限的实验技术难以区分,于是,过渡碳化物的真实结构成为了未解之谜.图11 [58 ] 所示的473 K下析出模拟结果显示出ε →η 的析出序列,表明这2种结构都会出现.图12 [58 ] 显示出不同温度下热力学驱动力、动力学能垒和析出组织间的相互关联:随着析出进行,热力学驱动力不断减小,动力学能垒不断增大;当温度提升后,析出物长大到同一尺寸时对应的热力学驱动力增大,动力学能垒减小.这说明热力学驱动力和动力学能垒以此消彼长的变化形式(即热-动力学相关性)来协同控制析出反应. ...
... [58 ]所示的473 K下析出模拟结果显示出ε →η 的析出序列,表明这2种结构都会出现.图12 [58 ] 显示出不同温度下热力学驱动力、动力学能垒和析出组织间的相互关联:随着析出进行,热力学驱动力不断减小,动力学能垒不断增大;当温度提升后,析出物长大到同一尺寸时对应的热力学驱动力增大,动力学能垒减小.这说明热力学驱动力和动力学能垒以此消彼长的变化形式(即热-动力学相关性)来协同控制析出反应. ...
... [58 ]显示出不同温度下热力学驱动力、动力学能垒和析出组织间的相互关联:随着析出进行,热力学驱动力不断减小,动力学能垒不断增大;当温度提升后,析出物长大到同一尺寸时对应的热力学驱动力增大,动力学能垒减小.这说明热力学驱动力和动力学能垒以此消彼长的变化形式(即热-动力学相关性)来协同控制析出反应. ...
... [
58 ]
Evolution for the energy of system and the number of primary building units (PBUs) in carbides during precipitations of <i>ε</i>-Fe<sub>2</sub>C, <i>η</i>-Fe<sub>2</sub>C, and <i>θ</i>-Fe<sub>3</sub>C in Fe-2%C (atomic fraction) alloy precipitated at 473 K (<i>τ</i><sub>1</sub> represents the moment for <i>ε</i>-Fe<sub>2</sub>C→<i>η</i>-Fe<sub>2</sub>C transition corresponding to the point o, and <i>τ</i><sub>2</sub> represents the time needed for <i>η</i>-Fe<sub>2</sub>C to grow to the same size as <i>ε</i>-Fe<sub>2</sub>C corresponding to the points F and Z)<sup>[<xref ref-type="bibr" rid="R58">58</xref>]</sup> Fig.11 ![]()
图12 <i>ε</i>-Fe<sub>2</sub>C和<i>η</i>-Fe<sub>2</sub>C析出时热力学驱动力和动力学能垒随着析出物中基础构成单元数量和温度的变化<sup>[<xref ref-type="bibr" rid="R58">58</xref>]</sup> Variations of the driving force (thermodynamics) and energy barrier (kinetics) with respect to the number of PBUs in precipitates and temperature for precipitations of <i>ε</i>-Fe<sub>2</sub>C and <i>η</i>-Fe<sub>2</sub>C<sup>[<xref ref-type="bibr" rid="R58">58</xref>]</sup> Fig.12 ![]()
相比相场法,该模型可以直接得到析出物结构参量演化,但由于选择有限个参量来描述微粒形貌,只能处理简单几何形状.此外,Fokker-Planck方程计算量偏大,推广性和可迁移性尚且不足.虽然如此,模型通过考虑相界面处原子跃迁这一促成析出反应的基本微观现象,从原子尺度计算出发,赋予多尺度模拟物理意义与可操作性,在不同金属材料中已呈现出应用价值.本团队针对现有问题,正对模型进行不断修正和检验,以使热-动力学协同的多尺度模拟更好地辅助工业复杂合金的理性设计. ...
... [
58 ]
Fig.11 ![]()
图12 <i>ε</i>-Fe<sub>2</sub>C和<i>η</i>-Fe<sub>2</sub>C析出时热力学驱动力和动力学能垒随着析出物中基础构成单元数量和温度的变化<sup>[<xref ref-type="bibr" rid="R58">58</xref>]</sup> Variations of the driving force (thermodynamics) and energy barrier (kinetics) with respect to the number of PBUs in precipitates and temperature for precipitations of <i>ε</i>-Fe<sub>2</sub>C and <i>η</i>-Fe<sub>2</sub>C<sup>[<xref ref-type="bibr" rid="R58">58</xref>]</sup> Fig.12 ![]()
相比相场法,该模型可以直接得到析出物结构参量演化,但由于选择有限个参量来描述微粒形貌,只能处理简单几何形状.此外,Fokker-Planck方程计算量偏大,推广性和可迁移性尚且不足.虽然如此,模型通过考虑相界面处原子跃迁这一促成析出反应的基本微观现象,从原子尺度计算出发,赋予多尺度模拟物理意义与可操作性,在不同金属材料中已呈现出应用价值.本团队针对现有问题,正对模型进行不断修正和检验,以使热-动力学协同的多尺度模拟更好地辅助工业复杂合金的理性设计. ...
... [
58 ]
Variations of the driving force (thermodynamics) and energy barrier (kinetics) with respect to the number of PBUs in precipitates and temperature for precipitations of <i>ε</i>-Fe<sub>2</sub>C and <i>η</i>-Fe<sub>2</sub>C<sup>[<xref ref-type="bibr" rid="R58">58</xref>]</sup> Fig.12 ![]()
相比相场法,该模型可以直接得到析出物结构参量演化,但由于选择有限个参量来描述微粒形貌,只能处理简单几何形状.此外,Fokker-Planck方程计算量偏大,推广性和可迁移性尚且不足.虽然如此,模型通过考虑相界面处原子跃迁这一促成析出反应的基本微观现象,从原子尺度计算出发,赋予多尺度模拟物理意义与可操作性,在不同金属材料中已呈现出应用价值.本团队针对现有问题,正对模型进行不断修正和检验,以使热-动力学协同的多尺度模拟更好地辅助工业复杂合金的理性设计. ...
... [
58 ]
Fig.12 ![]()
相比相场法,该模型可以直接得到析出物结构参量演化,但由于选择有限个参量来描述微粒形貌,只能处理简单几何形状.此外,Fokker-Planck方程计算量偏大,推广性和可迁移性尚且不足.虽然如此,模型通过考虑相界面处原子跃迁这一促成析出反应的基本微观现象,从原子尺度计算出发,赋予多尺度模拟物理意义与可操作性,在不同金属材料中已呈现出应用价值.本团队针对现有问题,正对模型进行不断修正和检验,以使热-动力学协同的多尺度模拟更好地辅助工业复杂合金的理性设计. ...
Modelling the overlap of nucleation, growth and coarsening during precipitation
1
2004
... KWN模型已成为Thermal-Calc软件中TC-PRISMA模块的核心理论,被广泛应用于复杂工业合金的析出相模拟、析出机理探究与经典理论发展.微粒尺寸分布,即一定尺寸范围内的析出物数量,对金属性能有很大影响.Robson[48 ] 借助KWN模型模拟了Cu-1.02%Co (原子分数)合金600℃下的析出反应,结果表明早期存在着晶核尺寸的双峰分布,是由形核与长大速率变化引起的,该现象也在其他实验与模拟中发现[49 ,56 ~58 ] .Robson[59 ] 还研究了形核、长大、粗化阶段出现的重叠现象,结果表明重叠程度与界面能、溶质过饱和度有很大关联而与溶质扩散性无关. ...
Simulation of precipitates evolution in multiphase multicomponent systems with consideration of nucleation
1
2016
... (2) 扩散场胞法.前述模型虽被广泛应用于不同情景,但均未考虑元素在基体中的扩散交互作用及不同析出物扩散场的重叠作用.这2种效应对析出动力学均会造成影响,Du等[51 ] 采用热力学极值原理构建出析出物演化模型,证实了考虑软碰撞的重要性.本小节介绍Popov等[60 ~62 ] 提出的同时考虑这2种效应的新模型(此处称作“扩散场胞法”). ...
Simulation of the evolution of precipitates in multicomponent alloys
2003
Simulation of VC precipitate evolution in steels with consideration for the formation of new nuclei
1
2005
... (2) 扩散场胞法.前述模型虽被广泛应用于不同情景,但均未考虑元素在基体中的扩散交互作用及不同析出物扩散场的重叠作用.这2种效应对析出动力学均会造成影响,Du等[51 ] 采用热力学极值原理构建出析出物演化模型,证实了考虑软碰撞的重要性.本小节介绍Popov等[60 ~62 ] 提出的同时考虑这2种效应的新模型(此处称作“扩散场胞法”). ...
Modified evolution equations for the precipitation kinetics of complex phases in multi-component systems
1
2004
... 对于复杂合金体系,相比偏微分扩散方程,由热力学极值原理得到的演化方程求解通常更为简单.Svoboda等[28 ] 和Kozeschnik等[29 ,63 ] 推导出控制多组分多相复杂合金体系下微粒析出演化的SFFK (Svoboda-Fischer-Fratzl-Kozeschnik)模型.对含有n 种组分(n =n s +n k ,n s 和n k 分别为置换和间隙原子数量)的合金体系,Ni 表示i 组元在体系中总数量(i =1, 2, ⋯ n ),Rp 表示析出相p 的半径(p =1, 2, ⋯ m ),p ci i 组元在相p 的平均浓度,γp 表示析出相p 的界面能,0 Ni 表示基体中i 组元数量,0 μi 表示i 组元在基体中化学势,p μi i 组元在相p 中化学势,则多种晶核析出时体系自由能可表示为: ...
1
2001
... 尽管SFFK模型中仍存在一些可调的未知参量(例如γp 、p Di Mp 、λ p 等),其已被植入到Matcalc软件[64 ] 中,成为除TC-PRISMA(即KWN模型)外又一广泛应用的析出相模拟工具[65 ~67 ] .图6 [29 ] 为采用不同界面能得到的600℃下Fe-Mn-Si-Cr-Mo-Ti-C体系中各种Mx Cy M 2 C),因此构建可靠、准确的热-动力学数据库对于析出模拟至关重要. ...
Precipitation behaviour of a complex steel
1
2006
... 尽管SFFK模型中仍存在一些可调的未知参量(例如γp 、p Di Mp 、λ p 等),其已被植入到Matcalc软件[64 ] 中,成为除TC-PRISMA(即KWN模型)外又一广泛应用的析出相模拟工具[65 ~67 ] .图6 [29 ] 为采用不同界面能得到的600℃下Fe-Mn-Si-Cr-Mo-Ti-C体系中各种Mx Cy M 2 C),因此构建可靠、准确的热-动力学数据库对于析出模拟至关重要. ...
Modeling precipitation thermodynamics and kinetics in type 316 austenitic stainless steels with varying composition as an initial step toward predicting phase stability during irradiation
2015
Mechanical property change and precipitate evolution during long-term aging of 1.25Cr-0.5Mo steel
1
2020
... 尽管SFFK模型中仍存在一些可调的未知参量(例如γp 、p Di Mp 、λ p 等),其已被植入到Matcalc软件[64 ] 中,成为除TC-PRISMA(即KWN模型)外又一广泛应用的析出相模拟工具[65 ~67 ] .图6 [29 ] 为采用不同界面能得到的600℃下Fe-Mn-Si-Cr-Mo-Ti-C体系中各种Mx Cy M 2 C),因此构建可靠、准确的热-动力学数据库对于析出模拟至关重要. ...
Microstructure-sensitive computational structure-property relations in materials design
1
2018
... CALPHAD数据库中相平衡数据比热化学数据更多一些,原因在于前者更易从实验获得[31 ] .随着计算材料学发展,第一性原理计算已成为获取热力学数据的有效途径.第一性原理可与CALPHAD计算以统计热力学为桥梁相结合,一方面补充CALPHAD中缺乏的数据,另一方面提升模型的预测性能.由于相变时材料结构的多时空间尺度变化[68 ] ,多尺度组织模拟对于理解与每个尺度下相关的性能变化至关重要,从第一性原理原子尺度出发的计算便为多尺度模拟提供了可能.本节简述第一性原理计算在析出相研究中的应用,分别介绍基于第一性原理计算的多尺度相场法模拟和基于Fokker-Planck方程的多尺度模拟. ...
High-throughput computing for accelerated materials discovery
1
2018
... 大规模并行的高通量计算常被用于挖掘材料的潜在性质[69 ] ,而基于密度泛函理论(density functional theory,DFT)的第一性原理计算是材料设计工程中高通量计算方法的首选.现有的DFT软件可提供合适的参数设置来保证计算精度,包括:Vienna ab initio simulation package (VASP)[70 ] 、Quantum Espresso[71 ] 、ABINIT[72 ] 等. ...
Ab-initio simulations of materials using VASP: Density-functional theory and beyond
1
2008
... 大规模并行的高通量计算常被用于挖掘材料的潜在性质[69 ] ,而基于密度泛函理论(density functional theory,DFT)的第一性原理计算是材料设计工程中高通量计算方法的首选.现有的DFT软件可提供合适的参数设置来保证计算精度,包括:Vienna ab initio simulation package (VASP)[70 ] 、Quantum Espresso[71 ] 、ABINIT[72 ] 等. ...
QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials
1
2009
... 大规模并行的高通量计算常被用于挖掘材料的潜在性质[69 ] ,而基于密度泛函理论(density functional theory,DFT)的第一性原理计算是材料设计工程中高通量计算方法的首选.现有的DFT软件可提供合适的参数设置来保证计算精度,包括:Vienna ab initio simulation package (VASP)[70 ] 、Quantum Espresso[71 ] 、ABINIT[72 ] 等. ...
ABINIT: First-principles approach to material and nanosystem properties
1
2009
... 大规模并行的高通量计算常被用于挖掘材料的潜在性质[69 ] ,而基于密度泛函理论(density functional theory,DFT)的第一性原理计算是材料设计工程中高通量计算方法的首选.现有的DFT软件可提供合适的参数设置来保证计算精度,包括:Vienna ab initio simulation package (VASP)[70 ] 、Quantum Espresso[71 ] 、ABINIT[72 ] 等. ...
Structure and stability of Fe2 C phases from density-functional theory calculations
1
2010
... DFT在析出热-动力学研究中发挥了重要作用,可以钢铁回火析出相(Fe2 C、M 3 C、M 7 C3 、M 23 C6 等)为例.通常,以析出物在0 K下的形成焓为热力学稳定性判据,可推断析出演化序列.基于此,Fang等[73 ] 通过比较回火过渡碳化物ε 和η 的形成焓推断出ε →η 的析出序列,该现象被相关实验[74 ] 和模拟[58 ] 所证实.形成焓也可反映成分对析出物形成的影响.Medvedeva等[75 ] 的形成焓计算表明,W和Mo的添加会使M 23 C6 更加稳定并减少其与基体间的晶格错配而有利于析出.Konyaeva等[76 ] 计算了(Fe, Cr)3 C和(Fe, Cr)7 C3 在不同置换原子浓度下的形成焓,判断出杂质原子在晶格上的优先占据位置.Ande等[77 ] 计算了合金元素在Fe3 C和铁素体间的配分焓,总结出合金元素对析出物成分变化的影响规律.利用DFT计算析出物力学性质可判断其机械稳定性.Liu等[78 ] 用应力应变法和Voigt-Reuss-Hill近似法,分别得到M 23 C6 的弹性常数和模量,表明有Fe或Mo掺杂的M 23 C6 比纯Cr23 C6 和Mn23 C6 的力学性能更好. ...
The ε →η →θ transition in 100Cr6 and its effect on mechanical properties
1
2012
... DFT在析出热-动力学研究中发挥了重要作用,可以钢铁回火析出相(Fe2 C、M 3 C、M 7 C3 、M 23 C6 等)为例.通常,以析出物在0 K下的形成焓为热力学稳定性判据,可推断析出演化序列.基于此,Fang等[73 ] 通过比较回火过渡碳化物ε 和η 的形成焓推断出ε →η 的析出序列,该现象被相关实验[74 ] 和模拟[58 ] 所证实.形成焓也可反映成分对析出物形成的影响.Medvedeva等[75 ] 的形成焓计算表明,W和Mo的添加会使M 23 C6 更加稳定并减少其与基体间的晶格错配而有利于析出.Konyaeva等[76 ] 计算了(Fe, Cr)3 C和(Fe, Cr)7 C3 在不同置换原子浓度下的形成焓,判断出杂质原子在晶格上的优先占据位置.Ande等[77 ] 计算了合金元素在Fe3 C和铁素体间的配分焓,总结出合金元素对析出物成分变化的影响规律.利用DFT计算析出物力学性质可判断其机械稳定性.Liu等[78 ] 用应力应变法和Voigt-Reuss-Hill近似法,分别得到M 23 C6 的弹性常数和模量,表明有Fe或Mo掺杂的M 23 C6 比纯Cr23 C6 和Mn23 C6 的力学性能更好. ...
Stability of binary and ternary M 23 C6 carbides from first principles
1
2015
... DFT在析出热-动力学研究中发挥了重要作用,可以钢铁回火析出相(Fe2 C、M 3 C、M 7 C3 、M 23 C6 等)为例.通常,以析出物在0 K下的形成焓为热力学稳定性判据,可推断析出演化序列.基于此,Fang等[73 ] 通过比较回火过渡碳化物ε 和η 的形成焓推断出ε →η 的析出序列,该现象被相关实验[74 ] 和模拟[58 ] 所证实.形成焓也可反映成分对析出物形成的影响.Medvedeva等[75 ] 的形成焓计算表明,W和Mo的添加会使M 23 C6 更加稳定并减少其与基体间的晶格错配而有利于析出.Konyaeva等[76 ] 计算了(Fe, Cr)3 C和(Fe, Cr)7 C3 在不同置换原子浓度下的形成焓,判断出杂质原子在晶格上的优先占据位置.Ande等[77 ] 计算了合金元素在Fe3 C和铁素体间的配分焓,总结出合金元素对析出物成分变化的影响规律.利用DFT计算析出物力学性质可判断其机械稳定性.Liu等[78 ] 用应力应变法和Voigt-Reuss-Hill近似法,分别得到M 23 C6 的弹性常数和模量,表明有Fe或Mo掺杂的M 23 C6 比纯Cr23 C6 和Mn23 C6 的力学性能更好. ...
Electronic structure, magnetic properties, and stability of the binary and ternary carbides (Fe, Cr)3 C and (Fe, Cr)7 C3
1
2009
... DFT在析出热-动力学研究中发挥了重要作用,可以钢铁回火析出相(Fe2 C、M 3 C、M 7 C3 、M 23 C6 等)为例.通常,以析出物在0 K下的形成焓为热力学稳定性判据,可推断析出演化序列.基于此,Fang等[73 ] 通过比较回火过渡碳化物ε 和η 的形成焓推断出ε →η 的析出序列,该现象被相关实验[74 ] 和模拟[58 ] 所证实.形成焓也可反映成分对析出物形成的影响.Medvedeva等[75 ] 的形成焓计算表明,W和Mo的添加会使M 23 C6 更加稳定并减少其与基体间的晶格错配而有利于析出.Konyaeva等[76 ] 计算了(Fe, Cr)3 C和(Fe, Cr)7 C3 在不同置换原子浓度下的形成焓,判断出杂质原子在晶格上的优先占据位置.Ande等[77 ] 计算了合金元素在Fe3 C和铁素体间的配分焓,总结出合金元素对析出物成分变化的影响规律.利用DFT计算析出物力学性质可判断其机械稳定性.Liu等[78 ] 用应力应变法和Voigt-Reuss-Hill近似法,分别得到M 23 C6 的弹性常数和模量,表明有Fe或Mo掺杂的M 23 C6 比纯Cr23 C6 和Mn23 C6 的力学性能更好. ...
First-principles prediction of partitioning of alloying elements between cementite and ferrite
1
2010
... DFT在析出热-动力学研究中发挥了重要作用,可以钢铁回火析出相(Fe2 C、M 3 C、M 7 C3 、M 23 C6 等)为例.通常,以析出物在0 K下的形成焓为热力学稳定性判据,可推断析出演化序列.基于此,Fang等[73 ] 通过比较回火过渡碳化物ε 和η 的形成焓推断出ε →η 的析出序列,该现象被相关实验[74 ] 和模拟[58 ] 所证实.形成焓也可反映成分对析出物形成的影响.Medvedeva等[75 ] 的形成焓计算表明,W和Mo的添加会使M 23 C6 更加稳定并减少其与基体间的晶格错配而有利于析出.Konyaeva等[76 ] 计算了(Fe, Cr)3 C和(Fe, Cr)7 C3 在不同置换原子浓度下的形成焓,判断出杂质原子在晶格上的优先占据位置.Ande等[77 ] 计算了合金元素在Fe3 C和铁素体间的配分焓,总结出合金元素对析出物成分变化的影响规律.利用DFT计算析出物力学性质可判断其机械稳定性.Liu等[78 ] 用应力应变法和Voigt-Reuss-Hill近似法,分别得到M 23 C6 的弹性常数和模量,表明有Fe或Mo掺杂的M 23 C6 比纯Cr23 C6 和Mn23 C6 的力学性能更好. ...
Mechanical properties and electronic structures of M 23 C6 (M =Fe, Cr, Mn)-type multicomponent carbides
1
2015
... DFT在析出热-动力学研究中发挥了重要作用,可以钢铁回火析出相(Fe2 C、M 3 C、M 7 C3 、M 23 C6 等)为例.通常,以析出物在0 K下的形成焓为热力学稳定性判据,可推断析出演化序列.基于此,Fang等[73 ] 通过比较回火过渡碳化物ε 和η 的形成焓推断出ε →η 的析出序列,该现象被相关实验[74 ] 和模拟[58 ] 所证实.形成焓也可反映成分对析出物形成的影响.Medvedeva等[75 ] 的形成焓计算表明,W和Mo的添加会使M 23 C6 更加稳定并减少其与基体间的晶格错配而有利于析出.Konyaeva等[76 ] 计算了(Fe, Cr)3 C和(Fe, Cr)7 C3 在不同置换原子浓度下的形成焓,判断出杂质原子在晶格上的优先占据位置.Ande等[77 ] 计算了合金元素在Fe3 C和铁素体间的配分焓,总结出合金元素对析出物成分变化的影响规律.利用DFT计算析出物力学性质可判断其机械稳定性.Liu等[78 ] 用应力应变法和Voigt-Reuss-Hill近似法,分别得到M 23 C6 的弹性常数和模量,表明有Fe或Mo掺杂的M 23 C6 比纯Cr23 C6 和Mn23 C6 的力学性能更好. ...
Cluster expansions and the configurational energy of alloys
1
1993
... 然而,以上应用都基于0 K,实际析出发生在有限温度下.有些析出物的基态不稳定,但可能因有限温度下的声子和电子激发变得稳定,因此有限温度下的热力学性能获取很有必要.有限温度下的自由能计算需考虑以下部分的贡献:0 K下的内能;原子尺度下各组分混合的构型熵;晶格振动对熵的贡献;电子热激发和自旋极化对熵的贡献[31 ] .对于理想混合,可直接由(-R Σxi lnxi )得到构型熵(xi 为i 组分摩尔分数),若存在短程有序,可用团簇扩张(cluster expansion)和Monte Carlo模拟[79 ,80 ] 来处理.通过简谐或准简谐近似可从DFT计算中获得声子谱,继而得到振动熵[31 ] .Wang等[81 ,82 ] 通过考虑有限温度下不同磁性状态的混合得到电子尺度的熵贡献.同样以碳化物为例.Fang等[83 ] 计算了Fe3 C和Fe2 C间相对自由能差随温度的变化,结果显示,当温度升高到330 K,稳定结构从Fe2 C变为Fe3 C,这与实验结果吻合,并说明Fe3 C的普遍存在由晶格振动和磁序变化所致.Kaplan等[84 ] 计算了Cr7 C3 的2种晶格结构的相对稳定性随温度的变化,表明低温条件下Cr7 C3 是六方结构(P 63 mc ),当温度升到470 K,会向正交结构(Pnma )转变.从以上热力学角度只可做初步分析,界面能和应变能会对析出过程产生很大影响,因此,考虑热-动力学协同的模拟会更有价值. ...
Self-driven lattice-model Monte Carlo simulations of alloy thermodynamic properties and phase diagrams
1
2002
... 然而,以上应用都基于0 K,实际析出发生在有限温度下.有些析出物的基态不稳定,但可能因有限温度下的声子和电子激发变得稳定,因此有限温度下的热力学性能获取很有必要.有限温度下的自由能计算需考虑以下部分的贡献:0 K下的内能;原子尺度下各组分混合的构型熵;晶格振动对熵的贡献;电子热激发和自旋极化对熵的贡献[31 ] .对于理想混合,可直接由(-R Σxi lnxi )得到构型熵(xi 为i 组分摩尔分数),若存在短程有序,可用团簇扩张(cluster expansion)和Monte Carlo模拟[79 ,80 ] 来处理.通过简谐或准简谐近似可从DFT计算中获得声子谱,继而得到振动熵[31 ] .Wang等[81 ,82 ] 通过考虑有限温度下不同磁性状态的混合得到电子尺度的熵贡献.同样以碳化物为例.Fang等[83 ] 计算了Fe3 C和Fe2 C间相对自由能差随温度的变化,结果显示,当温度升高到330 K,稳定结构从Fe2 C变为Fe3 C,这与实验结果吻合,并说明Fe3 C的普遍存在由晶格振动和磁序变化所致.Kaplan等[84 ] 计算了Cr7 C3 的2种晶格结构的相对稳定性随温度的变化,表明低温条件下Cr7 C3 是六方结构(P 63 mc ),当温度升到470 K,会向正交结构(Pnma )转变.从以上热力学角度只可做初步分析,界面能和应变能会对析出过程产生很大影响,因此,考虑热-动力学协同的模拟会更有价值. ...
Thermodynamics of the Ce γ -α transition: Density-functional study
1
2008
... 然而,以上应用都基于0 K,实际析出发生在有限温度下.有些析出物的基态不稳定,但可能因有限温度下的声子和电子激发变得稳定,因此有限温度下的热力学性能获取很有必要.有限温度下的自由能计算需考虑以下部分的贡献:0 K下的内能;原子尺度下各组分混合的构型熵;晶格振动对熵的贡献;电子热激发和自旋极化对熵的贡献[31 ] .对于理想混合,可直接由(-R Σxi lnxi )得到构型熵(xi 为i 组分摩尔分数),若存在短程有序,可用团簇扩张(cluster expansion)和Monte Carlo模拟[79 ,80 ] 来处理.通过简谐或准简谐近似可从DFT计算中获得声子谱,继而得到振动熵[31 ] .Wang等[81 ,82 ] 通过考虑有限温度下不同磁性状态的混合得到电子尺度的熵贡献.同样以碳化物为例.Fang等[83 ] 计算了Fe3 C和Fe2 C间相对自由能差随温度的变化,结果显示,当温度升高到330 K,稳定结构从Fe2 C变为Fe3 C,这与实验结果吻合,并说明Fe3 C的普遍存在由晶格振动和磁序变化所致.Kaplan等[84 ] 计算了Cr7 C3 的2种晶格结构的相对稳定性随温度的变化,表明低温条件下Cr7 C3 是六方结构(P 63 mc ),当温度升到470 K,会向正交结构(Pnma )转变.从以上热力学角度只可做初步分析,界面能和应变能会对析出过程产生很大影响,因此,考虑热-动力学协同的模拟会更有价值. ...
A thermodynamic framework for a system with itinerant-electron magnetism
1
2009
... 然而,以上应用都基于0 K,实际析出发生在有限温度下.有些析出物的基态不稳定,但可能因有限温度下的声子和电子激发变得稳定,因此有限温度下的热力学性能获取很有必要.有限温度下的自由能计算需考虑以下部分的贡献:0 K下的内能;原子尺度下各组分混合的构型熵;晶格振动对熵的贡献;电子热激发和自旋极化对熵的贡献[31 ] .对于理想混合,可直接由(-R Σxi lnxi )得到构型熵(xi 为i 组分摩尔分数),若存在短程有序,可用团簇扩张(cluster expansion)和Monte Carlo模拟[79 ,80 ] 来处理.通过简谐或准简谐近似可从DFT计算中获得声子谱,继而得到振动熵[31 ] .Wang等[81 ,82 ] 通过考虑有限温度下不同磁性状态的混合得到电子尺度的熵贡献.同样以碳化物为例.Fang等[83 ] 计算了Fe3 C和Fe2 C间相对自由能差随温度的变化,结果显示,当温度升高到330 K,稳定结构从Fe2 C变为Fe3 C,这与实验结果吻合,并说明Fe3 C的普遍存在由晶格振动和磁序变化所致.Kaplan等[84 ] 计算了Cr7 C3 的2种晶格结构的相对稳定性随温度的变化,表明低温条件下Cr7 C3 是六方结构(P 63 mc ),当温度升到470 K,会向正交结构(Pnma )转变.从以上热力学角度只可做初步分析,界面能和应变能会对析出过程产生很大影响,因此,考虑热-动力学协同的模拟会更有价值. ...
Origin of predominance of cementite among iron carbides in steel at elevated temperature
1
2010
... 然而,以上应用都基于0 K,实际析出发生在有限温度下.有些析出物的基态不稳定,但可能因有限温度下的声子和电子激发变得稳定,因此有限温度下的热力学性能获取很有必要.有限温度下的自由能计算需考虑以下部分的贡献:0 K下的内能;原子尺度下各组分混合的构型熵;晶格振动对熵的贡献;电子热激发和自旋极化对熵的贡献[31 ] .对于理想混合,可直接由(-R Σxi lnxi )得到构型熵(xi 为i 组分摩尔分数),若存在短程有序,可用团簇扩张(cluster expansion)和Monte Carlo模拟[79 ,80 ] 来处理.通过简谐或准简谐近似可从DFT计算中获得声子谱,继而得到振动熵[31 ] .Wang等[81 ,82 ] 通过考虑有限温度下不同磁性状态的混合得到电子尺度的熵贡献.同样以碳化物为例.Fang等[83 ] 计算了Fe3 C和Fe2 C间相对自由能差随温度的变化,结果显示,当温度升高到330 K,稳定结构从Fe2 C变为Fe3 C,这与实验结果吻合,并说明Fe3 C的普遍存在由晶格振动和磁序变化所致.Kaplan等[84 ] 计算了Cr7 C3 的2种晶格结构的相对稳定性随温度的变化,表明低温条件下Cr7 C3 是六方结构(P 63 mc ),当温度升到470 K,会向正交结构(Pnma )转变.从以上热力学角度只可做初步分析,界面能和应变能会对析出过程产生很大影响,因此,考虑热-动力学协同的模拟会更有价值. ...
Blomqvist A
1
2013
... 然而,以上应用都基于0 K,实际析出发生在有限温度下.有些析出物的基态不稳定,但可能因有限温度下的声子和电子激发变得稳定,因此有限温度下的热力学性能获取很有必要.有限温度下的自由能计算需考虑以下部分的贡献:0 K下的内能;原子尺度下各组分混合的构型熵;晶格振动对熵的贡献;电子热激发和自旋极化对熵的贡献[31 ] .对于理想混合,可直接由(-R Σxi lnxi )得到构型熵(xi 为i 组分摩尔分数),若存在短程有序,可用团簇扩张(cluster expansion)和Monte Carlo模拟[79 ,80 ] 来处理.通过简谐或准简谐近似可从DFT计算中获得声子谱,继而得到振动熵[31 ] .Wang等[81 ,82 ] 通过考虑有限温度下不同磁性状态的混合得到电子尺度的熵贡献.同样以碳化物为例.Fang等[83 ] 计算了Fe3 C和Fe2 C间相对自由能差随温度的变化,结果显示,当温度升高到330 K,稳定结构从Fe2 C变为Fe3 C,这与实验结果吻合,并说明Fe3 C的普遍存在由晶格振动和磁序变化所致.Kaplan等[84 ] 计算了Cr7 C3 的2种晶格结构的相对稳定性随温度的变化,表明低温条件下Cr7 C3 是六方结构(P 63 mc ),当温度升到470 K,会向正交结构(Pnma )转变.从以上热力学角度只可做初步分析,界面能和应变能会对析出过程产生很大影响,因此,考虑热-动力学协同的模拟会更有价值. ...
Phase-field models for microstructure evolution
1
2002
... 相场法是模拟结构演化的连续场方法,“相”指序参量,用以描述局部位置的化学和结构(例如成分、非弹性应变、晶格取向)的非均匀性.通过定义体系的序参量ϕ ,在求解其随时间的变化后,便可追溯体系结构演化.序参量分为守恒与非守恒序参量[85 ] :前者指在区域内保持守恒的物理量,例如成分、温度等;后者用来区分不同的相结构,例如析出相和基体.遵循热力学第二定律,序参量总是会朝着使体系自由能不断减小的方向变化.析出体系的总自由能F (ϕ )为: ...
On spinodal decomposition
1
1961
... 式中,F bulk 为块体自由能,F int 为析出相/基体界面能,F elastic 为微粒析出产生的应变能,f local 为局部位置r 处的自由能密度,f grad 为与序参量变化相关的自由能梯度项.若选择成分c 和用来区分相结构的η i 为ϕ ,则c 与η i 的演化需分别遵循Cahn-Hilliard扩散方程[86 ] 与Allen-Cahn方程[87 ] : ...
A microscopic theory for antiphase boundary motion and its application to antiphase domain coarsening
1
1979
... 式中,F bulk 为块体自由能,F int 为析出相/基体界面能,F elastic 为微粒析出产生的应变能,f local 为局部位置r 处的自由能密度,f grad 为与序参量变化相关的自由能梯度项.若选择成分c 和用来区分相结构的η i 为ϕ ,则c 与η i 的演化需分别遵循Cahn-Hilliard扩散方程[86 ] 与Allen-Cahn方程[87 ] : ...
Phase field modeling of defects and deformation
1
2010
... 式中,M s 为溶质移动性,L' 为与非守恒序参量相关的动力学系数.与KWN模型和SFFK模型的尖锐界面假设不同,相场法采用弥散界面处理[88 ] ,具有一定值的序参量可表示相界面.从式(40)可见,因无需直接追踪相界面位置,相场法擅长处理复杂析出物形貌.关于介观与微观相场法模拟的细致介绍与应用可见文献[89 ,90 ],本节简述将其与第一性原理计算结合进行的多尺度析出模拟. ...
An introduction to phase-field modeling of microstructure evolution
1
2008
... 式中,M s 为溶质移动性,L' 为与非守恒序参量相关的动力学系数.与KWN模型和SFFK模型的尖锐界面假设不同,相场法采用弥散界面处理[88 ] ,具有一定值的序参量可表示相界面.从式(40)可见,因无需直接追踪相界面位置,相场法擅长处理复杂析出物形貌.关于介观与微观相场法模拟的细致介绍与应用可见文献[89 ,90 ],本节简述将其与第一性原理计算结合进行的多尺度析出模拟. ...
Phase-field models in materials science
1
2009
... 式中,M s 为溶质移动性,L' 为与非守恒序参量相关的动力学系数.与KWN模型和SFFK模型的尖锐界面假设不同,相场法采用弥散界面处理[88 ] ,具有一定值的序参量可表示相界面.从式(40)可见,因无需直接追踪相界面位置,相场法擅长处理复杂析出物形貌.关于介观与微观相场法模拟的细致介绍与应用可见文献[89 ,90 ],本节简述将其与第一性原理计算结合进行的多尺度析出模拟. ...
A generalized computational interface for combined thermodynamic and kinetic modeling
1
2011
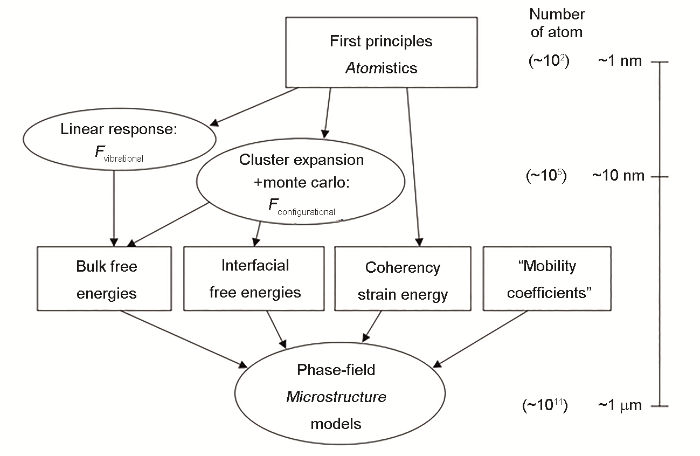
... 相场法模拟析出的准确度取决于对自由能式(39) 的表征精度.有2种方法可将CALPHAD与相场法结合以获取自由能参量:其一为直接与热力学数据库相连[91 ] ;其二为利用多项式近似CALPHAD自由能后再使用[92 ] .然而前者在求解时的每一步都需访问外部软件从而增大计算量,且商业软件需购买;后者在用多项式近似时难免损失热力学细节信息导致模拟准确性降低.如图7 [93 ] 所示,若从第一性原理计算出发分别求得块体自由能、界面能和应变能(式(39) ),则可完成从纳米级微观尺度到微米级介观尺度的多尺度模拟.Vaithyanathan等[93 ,94 ] 利用该多尺度框架模拟了铝合金中θ' -Al2 Cu的二维析出:将第一性原理计算、集团展开法和Monte Carlo法结合来得到Al基体的块体自由能;将第一性原理计算形成焓与振动熵结合得到θ' 的自由能;通过构建Al与θ' 的界面晶胞,由第一性原理得到在2个方向的半共格和共格界面能;通过第一性原理计算晶格常数和弹性常数等参量并代入Khachaturyan[95 ] 的解析表达式中得到应变能;最后利用所得自由能数据获取相场法中的参量(包含在式(39) 的具体表达形式中,为简化而略去),即可求解式(40)来完成模拟.图8 [94 ] 为Vaithyanathan等的结果,通过调节式(40b)中的L' 来选择是否考虑析出过程中的界面各向异性和弹性各向异性.可见,只有同时考虑这2种效应时,才能获得与实验吻合的θ' 片状形貌.Kim等[96 ] 在此基础上,进一步表达f grad 到四阶项,更精确地考虑了界面能各向异性、弹性各向异性、基体与析出物的弹性不均一性、错配应变各向异性,最终阐明了θ' 形貌的主导控制因素.因此相场法相比第2节中方法更适合研究控制析出过程的潜在物理机制. ...
A phase-field study of the aluminizing of nickel
1
2015
... 相场法模拟析出的准确度取决于对自由能式(39) 的表征精度.有2种方法可将CALPHAD与相场法结合以获取自由能参量:其一为直接与热力学数据库相连[91 ] ;其二为利用多项式近似CALPHAD自由能后再使用[92 ] .然而前者在求解时的每一步都需访问外部软件从而增大计算量,且商业软件需购买;后者在用多项式近似时难免损失热力学细节信息导致模拟准确性降低.如图7 [93 ] 所示,若从第一性原理计算出发分别求得块体自由能、界面能和应变能(式(39) ),则可完成从纳米级微观尺度到微米级介观尺度的多尺度模拟.Vaithyanathan等[93 ,94 ] 利用该多尺度框架模拟了铝合金中θ' -Al2 Cu的二维析出:将第一性原理计算、集团展开法和Monte Carlo法结合来得到Al基体的块体自由能;将第一性原理计算形成焓与振动熵结合得到θ' 的自由能;通过构建Al与θ' 的界面晶胞,由第一性原理得到在2个方向的半共格和共格界面能;通过第一性原理计算晶格常数和弹性常数等参量并代入Khachaturyan[95 ] 的解析表达式中得到应变能;最后利用所得自由能数据获取相场法中的参量(包含在式(39) 的具体表达形式中,为简化而略去),即可求解式(40)来完成模拟.图8 [94 ] 为Vaithyanathan等的结果,通过调节式(40b)中的L' 来选择是否考虑析出过程中的界面各向异性和弹性各向异性.可见,只有同时考虑这2种效应时,才能获得与实验吻合的θ' 片状形貌.Kim等[96 ] 在此基础上,进一步表达f grad 到四阶项,更精确地考虑了界面能各向异性、弹性各向异性、基体与析出物的弹性不均一性、错配应变各向异性,最终阐明了θ' 形貌的主导控制因素.因此相场法相比第2节中方法更适合研究控制析出过程的潜在物理机制. ...
Multiscale modeling of θ ′ precipitation in Al-Cu binary alloys
4
2004
... 相场法模拟析出的准确度取决于对自由能式(39) 的表征精度.有2种方法可将CALPHAD与相场法结合以获取自由能参量:其一为直接与热力学数据库相连[91 ] ;其二为利用多项式近似CALPHAD自由能后再使用[92 ] .然而前者在求解时的每一步都需访问外部软件从而增大计算量,且商业软件需购买;后者在用多项式近似时难免损失热力学细节信息导致模拟准确性降低.如图7 [93 ] 所示,若从第一性原理计算出发分别求得块体自由能、界面能和应变能(式(39) ),则可完成从纳米级微观尺度到微米级介观尺度的多尺度模拟.Vaithyanathan等[93 ,94 ] 利用该多尺度框架模拟了铝合金中θ' -Al2 Cu的二维析出:将第一性原理计算、集团展开法和Monte Carlo法结合来得到Al基体的块体自由能;将第一性原理计算形成焓与振动熵结合得到θ' 的自由能;通过构建Al与θ' 的界面晶胞,由第一性原理得到在2个方向的半共格和共格界面能;通过第一性原理计算晶格常数和弹性常数等参量并代入Khachaturyan[95 ] 的解析表达式中得到应变能;最后利用所得自由能数据获取相场法中的参量(包含在式(39) 的具体表达形式中,为简化而略去),即可求解式(40)来完成模拟.图8 [94 ] 为Vaithyanathan等的结果,通过调节式(40b)中的L' 来选择是否考虑析出过程中的界面各向异性和弹性各向异性.可见,只有同时考虑这2种效应时,才能获得与实验吻合的θ' 片状形貌.Kim等[96 ] 在此基础上,进一步表达f grad 到四阶项,更精确地考虑了界面能各向异性、弹性各向异性、基体与析出物的弹性不均一性、错配应变各向异性,最终阐明了θ' 形貌的主导控制因素.因此相场法相比第2节中方法更适合研究控制析出过程的潜在物理机制. ...
... [93 ,94 ]利用该多尺度框架模拟了铝合金中θ' -Al2 Cu的二维析出:将第一性原理计算、集团展开法和Monte Carlo法结合来得到Al基体的块体自由能;将第一性原理计算形成焓与振动熵结合得到θ' 的自由能;通过构建Al与θ' 的界面晶胞,由第一性原理得到在2个方向的半共格和共格界面能;通过第一性原理计算晶格常数和弹性常数等参量并代入Khachaturyan[95 ] 的解析表达式中得到应变能;最后利用所得自由能数据获取相场法中的参量(包含在式(39) 的具体表达形式中,为简化而略去),即可求解式(40)来完成模拟.图8 [94 ] 为Vaithyanathan等的结果,通过调节式(40b)中的L' 来选择是否考虑析出过程中的界面各向异性和弹性各向异性.可见,只有同时考虑这2种效应时,才能获得与实验吻合的θ' 片状形貌.Kim等[96 ] 在此基础上,进一步表达f grad 到四阶项,更精确地考虑了界面能各向异性、弹性各向异性、基体与析出物的弹性不均一性、错配应变各向异性,最终阐明了θ' 形貌的主导控制因素.因此相场法相比第2节中方法更适合研究控制析出过程的潜在物理机制. ...
... [
93 ]
Schematic of the multiscale model, showing different constituent models and their links along with their associated length scales (<i>F</i><sub>vibrational</sub>—free energy contributed from lattice vibration, <i>F</i><sub>configurational</sub>—configurational free energy)<sup>[<xref ref-type="bibr" rid="R93">93</xref>]</sup> Fig.7 ![]()
图8 在考虑不同各向异性条件下利用第一性原理所得热力学参量对<i>θ'</i>形貌的相场模拟<sup>[<xref ref-type="bibr" rid="R94">94</xref>]</sup> (a) isotropic (b) interface only (c) strain only ...
... [
93 ]
Fig.7 ![]()
图8 在考虑不同各向异性条件下利用第一性原理所得热力学参量对<i>θ'</i>形貌的相场模拟<sup>[<xref ref-type="bibr" rid="R94">94</xref>]</sup> (a) isotropic (b) interface only (c) strain only ...
Multiscale modeling of precipitate microstructure evolution
4
2002
... 相场法模拟析出的准确度取决于对自由能式(39) 的表征精度.有2种方法可将CALPHAD与相场法结合以获取自由能参量:其一为直接与热力学数据库相连[91 ] ;其二为利用多项式近似CALPHAD自由能后再使用[92 ] .然而前者在求解时的每一步都需访问外部软件从而增大计算量,且商业软件需购买;后者在用多项式近似时难免损失热力学细节信息导致模拟准确性降低.如图7 [93 ] 所示,若从第一性原理计算出发分别求得块体自由能、界面能和应变能(式(39) ),则可完成从纳米级微观尺度到微米级介观尺度的多尺度模拟.Vaithyanathan等[93 ,94 ] 利用该多尺度框架模拟了铝合金中θ' -Al2 Cu的二维析出:将第一性原理计算、集团展开法和Monte Carlo法结合来得到Al基体的块体自由能;将第一性原理计算形成焓与振动熵结合得到θ' 的自由能;通过构建Al与θ' 的界面晶胞,由第一性原理得到在2个方向的半共格和共格界面能;通过第一性原理计算晶格常数和弹性常数等参量并代入Khachaturyan[95 ] 的解析表达式中得到应变能;最后利用所得自由能数据获取相场法中的参量(包含在式(39) 的具体表达形式中,为简化而略去),即可求解式(40)来完成模拟.图8 [94 ] 为Vaithyanathan等的结果,通过调节式(40b)中的L' 来选择是否考虑析出过程中的界面各向异性和弹性各向异性.可见,只有同时考虑这2种效应时,才能获得与实验吻合的θ' 片状形貌.Kim等[96 ] 在此基础上,进一步表达f grad 到四阶项,更精确地考虑了界面能各向异性、弹性各向异性、基体与析出物的弹性不均一性、错配应变各向异性,最终阐明了θ' 形貌的主导控制因素.因此相场法相比第2节中方法更适合研究控制析出过程的潜在物理机制. ...
... [94 ]为Vaithyanathan等的结果,通过调节式(40b)中的L' 来选择是否考虑析出过程中的界面各向异性和弹性各向异性.可见,只有同时考虑这2种效应时,才能获得与实验吻合的θ' 片状形貌.Kim等[96 ] 在此基础上,进一步表达f grad 到四阶项,更精确地考虑了界面能各向异性、弹性各向异性、基体与析出物的弹性不均一性、错配应变各向异性,最终阐明了θ' 形貌的主导控制因素.因此相场法相比第2节中方法更适合研究控制析出过程的潜在物理机制. ...
... [
94 ]
(a) isotropic (b) interface only (c) strain only ...
... (d) interface+strain (e) interface+strain+kinetics (f) experiment
Phase-field simulation using thermodynamic parameters from first principles, showing <i>θ'</i> morphologies obtained with different anisotropic contributions<sup>[<xref ref-type="bibr" rid="R94">94</xref>]</sup> Fig.8 ![]()
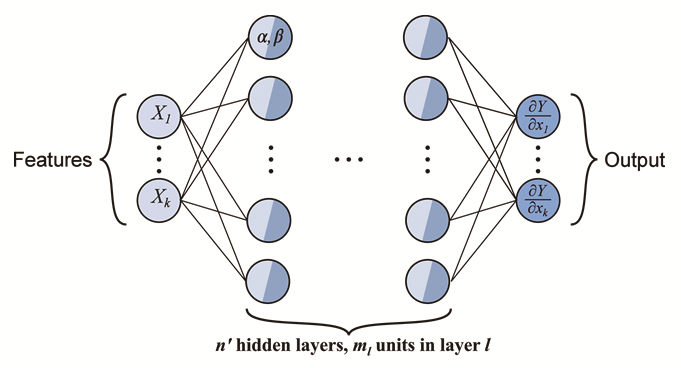
材料信息学中机器学习方法可充当第一性原理与相场法间联系的多尺度桥梁.Teichert等[97 ,98 ] 提出了一个用于获取相场法中自由能的主动学习(active learning)流程,并成功用于Ni3 Al的析出模拟.通过将第一性原理计算、团簇扩张法与Monte Carlo统计力学结合得到不同序参量组合中ηi 对应的共轭化学势(即∂F /∂ηi );然后以此为数据训练神经网络,如图9 [98 ] 所示,将序参量输入到最左边的输入层,不断调整中间隐藏层中参量后,最终输出与初始数据相差无几的共轭化学势,该过程实质为构建由序参量张成的自由能曲面;最后将训练好的神经网络与相场结合,每一步由神经网络为相场提供自由能数据便可完成模拟. ...
1
1983
... 相场法模拟析出的准确度取决于对自由能式(39) 的表征精度.有2种方法可将CALPHAD与相场法结合以获取自由能参量:其一为直接与热力学数据库相连[91 ] ;其二为利用多项式近似CALPHAD自由能后再使用[92 ] .然而前者在求解时的每一步都需访问外部软件从而增大计算量,且商业软件需购买;后者在用多项式近似时难免损失热力学细节信息导致模拟准确性降低.如图7 [93 ] 所示,若从第一性原理计算出发分别求得块体自由能、界面能和应变能(式(39) ),则可完成从纳米级微观尺度到微米级介观尺度的多尺度模拟.Vaithyanathan等[93 ,94 ] 利用该多尺度框架模拟了铝合金中θ' -Al2 Cu的二维析出:将第一性原理计算、集团展开法和Monte Carlo法结合来得到Al基体的块体自由能;将第一性原理计算形成焓与振动熵结合得到θ' 的自由能;通过构建Al与θ' 的界面晶胞,由第一性原理得到在2个方向的半共格和共格界面能;通过第一性原理计算晶格常数和弹性常数等参量并代入Khachaturyan[95 ] 的解析表达式中得到应变能;最后利用所得自由能数据获取相场法中的参量(包含在式(39) 的具体表达形式中,为简化而略去),即可求解式(40)来完成模拟.图8 [94 ] 为Vaithyanathan等的结果,通过调节式(40b)中的L' 来选择是否考虑析出过程中的界面各向异性和弹性各向异性.可见,只有同时考虑这2种效应时,才能获得与实验吻合的θ' 片状形貌.Kim等[96 ] 在此基础上,进一步表达f grad 到四阶项,更精确地考虑了界面能各向异性、弹性各向异性、基体与析出物的弹性不均一性、错配应变各向异性,最终阐明了θ' 形貌的主导控制因素.因此相场法相比第2节中方法更适合研究控制析出过程的潜在物理机制. ...
First-principles/phase-field modeling of θ ′ precipitation in Al-Cu alloys
1
2017
... 相场法模拟析出的准确度取决于对自由能式(39) 的表征精度.有2种方法可将CALPHAD与相场法结合以获取自由能参量:其一为直接与热力学数据库相连[91 ] ;其二为利用多项式近似CALPHAD自由能后再使用[92 ] .然而前者在求解时的每一步都需访问外部软件从而增大计算量,且商业软件需购买;后者在用多项式近似时难免损失热力学细节信息导致模拟准确性降低.如图7 [93 ] 所示,若从第一性原理计算出发分别求得块体自由能、界面能和应变能(式(39) ),则可完成从纳米级微观尺度到微米级介观尺度的多尺度模拟.Vaithyanathan等[93 ,94 ] 利用该多尺度框架模拟了铝合金中θ' -Al2 Cu的二维析出:将第一性原理计算、集团展开法和Monte Carlo法结合来得到Al基体的块体自由能;将第一性原理计算形成焓与振动熵结合得到θ' 的自由能;通过构建Al与θ' 的界面晶胞,由第一性原理得到在2个方向的半共格和共格界面能;通过第一性原理计算晶格常数和弹性常数等参量并代入Khachaturyan[95 ] 的解析表达式中得到应变能;最后利用所得自由能数据获取相场法中的参量(包含在式(39) 的具体表达形式中,为简化而略去),即可求解式(40)来完成模拟.图8 [94 ] 为Vaithyanathan等的结果,通过调节式(40b)中的L' 来选择是否考虑析出过程中的界面各向异性和弹性各向异性.可见,只有同时考虑这2种效应时,才能获得与实验吻合的θ' 片状形貌.Kim等[96 ] 在此基础上,进一步表达f grad 到四阶项,更精确地考虑了界面能各向异性、弹性各向异性、基体与析出物的弹性不均一性、错配应变各向异性,最终阐明了θ' 形貌的主导控制因素.因此相场法相比第2节中方法更适合研究控制析出过程的潜在物理机制. ...
Machine learning materials physics: Integrable deep neural networks enable scale bridging by learning free energy functions
1
2019
... 材料信息学中机器学习方法可充当第一性原理与相场法间联系的多尺度桥梁.Teichert等[97 ,98 ] 提出了一个用于获取相场法中自由能的主动学习(active learning)流程,并成功用于Ni3 Al的析出模拟.通过将第一性原理计算、团簇扩张法与Monte Carlo统计力学结合得到不同序参量组合中ηi 对应的共轭化学势(即∂F /∂ηi );然后以此为数据训练神经网络,如图9 [98 ] 所示,将序参量输入到最左边的输入层,不断调整中间隐藏层中参量后,最终输出与初始数据相差无几的共轭化学势,该过程实质为构建由序参量张成的自由能曲面;最后将训练好的神经网络与相场结合,每一步由神经网络为相场提供自由能数据便可完成模拟. ...
Scale bridging materials physics: Active learning workflows and integrable deep neural networks for free energy function representations in alloys
4
2020
... 材料信息学中机器学习方法可充当第一性原理与相场法间联系的多尺度桥梁.Teichert等[97 ,98 ] 提出了一个用于获取相场法中自由能的主动学习(active learning)流程,并成功用于Ni3 Al的析出模拟.通过将第一性原理计算、团簇扩张法与Monte Carlo统计力学结合得到不同序参量组合中ηi 对应的共轭化学势(即∂F /∂ηi );然后以此为数据训练神经网络,如图9 [98 ] 所示,将序参量输入到最左边的输入层,不断调整中间隐藏层中参量后,最终输出与初始数据相差无几的共轭化学势,该过程实质为构建由序参量张成的自由能曲面;最后将训练好的神经网络与相场结合,每一步由神经网络为相场提供自由能数据便可完成模拟. ...
... [98 ]所示,将序参量输入到最左边的输入层,不断调整中间隐藏层中参量后,最终输出与初始数据相差无几的共轭化学势,该过程实质为构建由序参量张成的自由能曲面;最后将训练好的神经网络与相场结合,每一步由神经网络为相场提供自由能数据便可完成模拟. ...
... [
98 ]
Schematic of an integrable deep neural network (<i>X<sub>k</sub></i> is the <i>k</i>-th input parameter, <i>Y</i> is the output of deep neural network, <span class="formulaText"><inline-formula><math id="M104"><mfrac><mrow><mo>∂</mo><mi>Y</mi></mrow><mrow><mo>∂</mo><msub><mrow><mi>X</mi></mrow><mrow><mi>k</mi></mrow></msub></mrow></mfrac></math></span></inline-formula></span> is the output of integrable deep neural network, <i>α</i> and <i>β</i> are adjustable parameters of the hidden layers)<sup>[<xref ref-type="bibr" rid="R98">98</xref>]</sup> Fig.9 ![]()
经典形核理论基于尖锐界面假设,将析出物视作球形,采用界面能各向同性近似,而这些假设均无需应用在相场法中.在考虑界面能和应变能各向异性后,利用相场法研究析出物形核现象有助于挖掘一些经典理论无法描述的新机理,譬如,Zhang等[99 ] 研究了弹性能和界面能各向异性对临界核胚形貌的影响.相场只朝着自由能减少的方向演化,但形核阶段会出现能量升高,可通过Langevin噪音法(Langevin noise method)[100 ] 与显式形核法(explicit nucleation method)[101 ] 解决该问题.前者在式(40)中加入噪声项来促使形核;后者利用经典形核理论计算每个位置处形核概率后,通过改变序参量来在任意时刻某一位置引入核胚.在有限体积分数下,当析出物间距离比较近时便会有扩散场交互作用,LSW粗化理论因只适用于小体积分数的情形而不再有效.Vaithyanathan等[102 ] 用相场法研究了δ' -Al3 Li与体积分数相关的粗化现象.此外,由于相场模型从自由能出发构建,各种应变场、磁场、电场等驱动力均可包含入相场中,使得相场模拟的物理意义更加明确.Li等[103 ] 便通过修改式(39) 研究了位错与施加应力对析出物形貌演化的影响,依此提出了调整工艺以控制组织形貌的方案.尽管相场法理论上可以处理任意复杂的形貌,但相比KWN模型等,由于考虑更加精细的微观结构,计算量一般偏大,体积分数、晶核尺寸分布、长大速率等参量需在形貌基础上进一步分析才能获得. ...
... [
98 ]
Fig.9 ![]()
经典形核理论基于尖锐界面假设,将析出物视作球形,采用界面能各向同性近似,而这些假设均无需应用在相场法中.在考虑界面能和应变能各向异性后,利用相场法研究析出物形核现象有助于挖掘一些经典理论无法描述的新机理,譬如,Zhang等[99 ] 研究了弹性能和界面能各向异性对临界核胚形貌的影响.相场只朝着自由能减少的方向演化,但形核阶段会出现能量升高,可通过Langevin噪音法(Langevin noise method)[100 ] 与显式形核法(explicit nucleation method)[101 ] 解决该问题.前者在式(40)中加入噪声项来促使形核;后者利用经典形核理论计算每个位置处形核概率后,通过改变序参量来在任意时刻某一位置引入核胚.在有限体积分数下,当析出物间距离比较近时便会有扩散场交互作用,LSW粗化理论因只适用于小体积分数的情形而不再有效.Vaithyanathan等[102 ] 用相场法研究了δ' -Al3 Li与体积分数相关的粗化现象.此外,由于相场模型从自由能出发构建,各种应变场、磁场、电场等驱动力均可包含入相场中,使得相场模拟的物理意义更加明确.Li等[103 ] 便通过修改式(39) 研究了位错与施加应力对析出物形貌演化的影响,依此提出了调整工艺以控制组织形貌的方案.尽管相场法理论上可以处理任意复杂的形貌,但相比KWN模型等,由于考虑更加精细的微观结构,计算量一般偏大,体积分数、晶核尺寸分布、长大速率等参量需在形貌基础上进一步分析才能获得. ...
Diffuse-interface description of strain-dominated morphology of critical nuclei in phase transformations
1
2008
... 经典形核理论基于尖锐界面假设,将析出物视作球形,采用界面能各向同性近似,而这些假设均无需应用在相场法中.在考虑界面能和应变能各向异性后,利用相场法研究析出物形核现象有助于挖掘一些经典理论无法描述的新机理,譬如,Zhang等[99 ] 研究了弹性能和界面能各向异性对临界核胚形貌的影响.相场只朝着自由能减少的方向演化,但形核阶段会出现能量升高,可通过Langevin噪音法(Langevin noise method)[100 ] 与显式形核法(explicit nucleation method)[101 ] 解决该问题.前者在式(40)中加入噪声项来促使形核;后者利用经典形核理论计算每个位置处形核概率后,通过改变序参量来在任意时刻某一位置引入核胚.在有限体积分数下,当析出物间距离比较近时便会有扩散场交互作用,LSW粗化理论因只适用于小体积分数的情形而不再有效.Vaithyanathan等[102 ] 用相场法研究了δ' -Al3 Li与体积分数相关的粗化现象.此外,由于相场模型从自由能出发构建,各种应变场、磁场、电场等驱动力均可包含入相场中,使得相场模拟的物理意义更加明确.Li等[103 ] 便通过修改式(39) 研究了位错与施加应力对析出物形貌演化的影响,依此提出了调整工艺以控制组织形貌的方案.尽管相场法理论上可以处理任意复杂的形貌,但相比KWN模型等,由于考虑更加精细的微观结构,计算量一般偏大,体积分数、晶核尺寸分布、长大速率等参量需在形貌基础上进一步分析才能获得. ...
Phase-field modeling of bimodal microstructures in nickel-based superalloys
1
2009
... 经典形核理论基于尖锐界面假设,将析出物视作球形,采用界面能各向同性近似,而这些假设均无需应用在相场法中.在考虑界面能和应变能各向异性后,利用相场法研究析出物形核现象有助于挖掘一些经典理论无法描述的新机理,譬如,Zhang等[99 ] 研究了弹性能和界面能各向异性对临界核胚形貌的影响.相场只朝着自由能减少的方向演化,但形核阶段会出现能量升高,可通过Langevin噪音法(Langevin noise method)[100 ] 与显式形核法(explicit nucleation method)[101 ] 解决该问题.前者在式(40)中加入噪声项来促使形核;后者利用经典形核理论计算每个位置处形核概率后,通过改变序参量来在任意时刻某一位置引入核胚.在有限体积分数下,当析出物间距离比较近时便会有扩散场交互作用,LSW粗化理论因只适用于小体积分数的情形而不再有效.Vaithyanathan等[102 ] 用相场法研究了δ' -Al3 Li与体积分数相关的粗化现象.此外,由于相场模型从自由能出发构建,各种应变场、磁场、电场等驱动力均可包含入相场中,使得相场模拟的物理意义更加明确.Li等[103 ] 便通过修改式(39) 研究了位错与施加应力对析出物形貌演化的影响,依此提出了调整工艺以控制组织形貌的方案.尽管相场法理论上可以处理任意复杂的形貌,但相比KWN模型等,由于考虑更加精细的微观结构,计算量一般偏大,体积分数、晶核尺寸分布、长大速率等参量需在形貌基础上进一步分析才能获得. ...
Phase field modeling of simultaneous nucleation and growth by explicitly incorporating nucleation events
1
2000
... 经典形核理论基于尖锐界面假设,将析出物视作球形,采用界面能各向同性近似,而这些假设均无需应用在相场法中.在考虑界面能和应变能各向异性后,利用相场法研究析出物形核现象有助于挖掘一些经典理论无法描述的新机理,譬如,Zhang等[99 ] 研究了弹性能和界面能各向异性对临界核胚形貌的影响.相场只朝着自由能减少的方向演化,但形核阶段会出现能量升高,可通过Langevin噪音法(Langevin noise method)[100 ] 与显式形核法(explicit nucleation method)[101 ] 解决该问题.前者在式(40)中加入噪声项来促使形核;后者利用经典形核理论计算每个位置处形核概率后,通过改变序参量来在任意时刻某一位置引入核胚.在有限体积分数下,当析出物间距离比较近时便会有扩散场交互作用,LSW粗化理论因只适用于小体积分数的情形而不再有效.Vaithyanathan等[102 ] 用相场法研究了δ' -Al3 Li与体积分数相关的粗化现象.此外,由于相场模型从自由能出发构建,各种应变场、磁场、电场等驱动力均可包含入相场中,使得相场模拟的物理意义更加明确.Li等[103 ] 便通过修改式(39) 研究了位错与施加应力对析出物形貌演化的影响,依此提出了调整工艺以控制组织形貌的方案.尽管相场法理论上可以处理任意复杂的形貌,但相比KWN模型等,由于考虑更加精细的微观结构,计算量一般偏大,体积分数、晶核尺寸分布、长大速率等参量需在形貌基础上进一步分析才能获得. ...
Coarsening kinetics of δ ′-Al3 Li precipitates: Phase-field simulation in 2D and 3D
1
2000
... 经典形核理论基于尖锐界面假设,将析出物视作球形,采用界面能各向同性近似,而这些假设均无需应用在相场法中.在考虑界面能和应变能各向异性后,利用相场法研究析出物形核现象有助于挖掘一些经典理论无法描述的新机理,譬如,Zhang等[99 ] 研究了弹性能和界面能各向异性对临界核胚形貌的影响.相场只朝着自由能减少的方向演化,但形核阶段会出现能量升高,可通过Langevin噪音法(Langevin noise method)[100 ] 与显式形核法(explicit nucleation method)[101 ] 解决该问题.前者在式(40)中加入噪声项来促使形核;后者利用经典形核理论计算每个位置处形核概率后,通过改变序参量来在任意时刻某一位置引入核胚.在有限体积分数下,当析出物间距离比较近时便会有扩散场交互作用,LSW粗化理论因只适用于小体积分数的情形而不再有效.Vaithyanathan等[102 ] 用相场法研究了δ' -Al3 Li与体积分数相关的粗化现象.此外,由于相场模型从自由能出发构建,各种应变场、磁场、电场等驱动力均可包含入相场中,使得相场模拟的物理意义更加明确.Li等[103 ] 便通过修改式(39) 研究了位错与施加应力对析出物形貌演化的影响,依此提出了调整工艺以控制组织形貌的方案.尽管相场法理论上可以处理任意复杂的形貌,但相比KWN模型等,由于考虑更加精细的微观结构,计算量一般偏大,体积分数、晶核尺寸分布、长大速率等参量需在形貌基础上进一步分析才能获得. ...
Phase field simulation of precipitates morphology with dislocations under applied stress
1
2011
... 经典形核理论基于尖锐界面假设,将析出物视作球形,采用界面能各向同性近似,而这些假设均无需应用在相场法中.在考虑界面能和应变能各向异性后,利用相场法研究析出物形核现象有助于挖掘一些经典理论无法描述的新机理,譬如,Zhang等[99 ] 研究了弹性能和界面能各向异性对临界核胚形貌的影响.相场只朝着自由能减少的方向演化,但形核阶段会出现能量升高,可通过Langevin噪音法(Langevin noise method)[100 ] 与显式形核法(explicit nucleation method)[101 ] 解决该问题.前者在式(40)中加入噪声项来促使形核;后者利用经典形核理论计算每个位置处形核概率后,通过改变序参量来在任意时刻某一位置引入核胚.在有限体积分数下,当析出物间距离比较近时便会有扩散场交互作用,LSW粗化理论因只适用于小体积分数的情形而不再有效.Vaithyanathan等[102 ] 用相场法研究了δ' -Al3 Li与体积分数相关的粗化现象.此外,由于相场模型从自由能出发构建,各种应变场、磁场、电场等驱动力均可包含入相场中,使得相场模拟的物理意义更加明确.Li等[103 ] 便通过修改式(39) 研究了位错与施加应力对析出物形貌演化的影响,依此提出了调整工艺以控制组织形貌的方案.尽管相场法理论上可以处理任意复杂的形貌,但相比KWN模型等,由于考虑更加精细的微观结构,计算量一般偏大,体积分数、晶核尺寸分布、长大速率等参量需在形貌基础上进一步分析才能获得. ...
Atomistic determination on stability, cluster and microstructures in terms of crystallographic and thermo-kinetic integration of Al-Mg-Si alloys
1
2020
... 该模型正确而可靠地模拟了铝合金的析出过程.对于Al-2%Cu (原子分数)合金[30 ] :基于晶体学信息构建出原子构型后,采用第一性原理和分子动力学模拟得到Φ ;可通过变分过渡态理论计算速率常数获得Mij ,为简化计算,用其与扩散系数间的关系6 D i a 2 S i * S i * i 组分原子数).通过交替方向隐式有限差分法求解式(41) 得到结论:图10 a[30 ] 中该模型所得θ' 尺寸随时间的变化(红、蓝、黑色线)比相场法结果(紫色线)更加吻合实验数据(绿、浅蓝色线);图10 b[30 ] 中的能量耗散路径体现出在实验中被大量证实的GP区→θ" →θ' 析出序列.类似地,模拟得到了与实验数据吻合的Al-Mg-Si合金中β" 析出动力学[104 ] . ...
Generalized stability criterion for exploiting optimized mechanical properties by a general correlation between phase transformations and plastic deformations
1
2020
... 最近,本团队通过提出“广义稳定性”的新概念将相变与变形一体化处理[105 ] .研究表明,相变中“大热力学驱动力-大动力学能垒-大广义稳定性”的对应关系会以组织结构为媒介被随后的变形过程所继承,变形中的“大驱动力”由极端非平衡工艺引发的位错交互作用引起,而“大能垒”会使大量位错运动得更为缓慢且绵长,从而推迟塑性失稳阶段的出现,在保证大强度的同时,实现了塑性的提升,这可以称之为,强塑性互斥关系在热/动力学协同作用下的突破.于是,“广义稳定性”指引下以性能为导向的模拟必然会辅助热-动力学协同的析出相模拟,一同助力贯通且关联“成分-工艺-组织-性能”的闭环,实现析出强化主导的高强高塑先进金属材料理性设计. ...





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}