Experimental investigations on the synthesis of W-Cu nanocomposite through spark plasma sintering
1
2015
... W-Cu材料由于其良好的力学性能和功能特性,在航空航天、电子信息、国防工业、电工和机械加工等领域[1,2,3,4,5,6]均有广泛运用.研究[7,8]表明,某些特定合金化元素添加可有效提高合金材料的使用性能和稳定性.例如,Chen等[9,10]研究了Fe、Co、Ni等元素对W-Cu粉末烧结过程的活化效果.结果表明,合金化元素的添加能够有效提高W在Cu液相中的溶解度,改善W和Cu的界面结合.Li等[11]在W-Cu体系中添加Ag元素,通过元素活化作用在较低烧结温度下获得了致密化良好的合金材料,且该体系具有良好的力学性能.Borji等[12]研究了合金化元素对W-Cu材料耐蚀性的影响,发现元素Ni可在促进W-Cu材料烧结致密化的同时增强体系的耐蚀性.Chookajorn等[13]在W体系中加入合金化元素Ti,制备了纳米晶W-Ti体系,其在1100 ℃的高温条件下仍能保持纳米晶晶粒组织,表明特定的合金化元素添加可显著增强纳米晶合金体系的热稳定性. ...
Infiltrated W-Cu composites with combined architecture of hierarchical particulate tungsten and tungsten fibers
1
2015
... W-Cu材料由于其良好的力学性能和功能特性,在航空航天、电子信息、国防工业、电工和机械加工等领域[1,2,3,4,5,6]均有广泛运用.研究[7,8]表明,某些特定合金化元素添加可有效提高合金材料的使用性能和稳定性.例如,Chen等[9,10]研究了Fe、Co、Ni等元素对W-Cu粉末烧结过程的活化效果.结果表明,合金化元素的添加能够有效提高W在Cu液相中的溶解度,改善W和Cu的界面结合.Li等[11]在W-Cu体系中添加Ag元素,通过元素活化作用在较低烧结温度下获得了致密化良好的合金材料,且该体系具有良好的力学性能.Borji等[12]研究了合金化元素对W-Cu材料耐蚀性的影响,发现元素Ni可在促进W-Cu材料烧结致密化的同时增强体系的耐蚀性.Chookajorn等[13]在W体系中加入合金化元素Ti,制备了纳米晶W-Ti体系,其在1100 ℃的高温条件下仍能保持纳米晶晶粒组织,表明特定的合金化元素添加可显著增强纳米晶合金体系的热稳定性. ...
A novel preparation method for W-Cu composite powders
1
2016
... W-Cu材料由于其良好的力学性能和功能特性,在航空航天、电子信息、国防工业、电工和机械加工等领域[1,2,3,4,5,6]均有广泛运用.研究[7,8]表明,某些特定合金化元素添加可有效提高合金材料的使用性能和稳定性.例如,Chen等[9,10]研究了Fe、Co、Ni等元素对W-Cu粉末烧结过程的活化效果.结果表明,合金化元素的添加能够有效提高W在Cu液相中的溶解度,改善W和Cu的界面结合.Li等[11]在W-Cu体系中添加Ag元素,通过元素活化作用在较低烧结温度下获得了致密化良好的合金材料,且该体系具有良好的力学性能.Borji等[12]研究了合金化元素对W-Cu材料耐蚀性的影响,发现元素Ni可在促进W-Cu材料烧结致密化的同时增强体系的耐蚀性.Chookajorn等[13]在W体系中加入合金化元素Ti,制备了纳米晶W-Ti体系,其在1100 ℃的高温条件下仍能保持纳米晶晶粒组织,表明特定的合金化元素添加可显著增强纳米晶合金体系的热稳定性. ...
Fabrication of W-Cu composite by shock consolidation of Cu-coated W powders
1
2016
... W-Cu材料由于其良好的力学性能和功能特性,在航空航天、电子信息、国防工业、电工和机械加工等领域[1,2,3,4,5,6]均有广泛运用.研究[7,8]表明,某些特定合金化元素添加可有效提高合金材料的使用性能和稳定性.例如,Chen等[9,10]研究了Fe、Co、Ni等元素对W-Cu粉末烧结过程的活化效果.结果表明,合金化元素的添加能够有效提高W在Cu液相中的溶解度,改善W和Cu的界面结合.Li等[11]在W-Cu体系中添加Ag元素,通过元素活化作用在较低烧结温度下获得了致密化良好的合金材料,且该体系具有良好的力学性能.Borji等[12]研究了合金化元素对W-Cu材料耐蚀性的影响,发现元素Ni可在促进W-Cu材料烧结致密化的同时增强体系的耐蚀性.Chookajorn等[13]在W体系中加入合金化元素Ti,制备了纳米晶W-Ti体系,其在1100 ℃的高温条件下仍能保持纳米晶晶粒组织,表明特定的合金化元素添加可显著增强纳米晶合金体系的热稳定性. ...
Microstructure and processing of 3D printed tungsten microlattices and infiltrated W-Cu composites
1
2018
... W-Cu材料由于其良好的力学性能和功能特性,在航空航天、电子信息、国防工业、电工和机械加工等领域[1,2,3,4,5,6]均有广泛运用.研究[7,8]表明,某些特定合金化元素添加可有效提高合金材料的使用性能和稳定性.例如,Chen等[9,10]研究了Fe、Co、Ni等元素对W-Cu粉末烧结过程的活化效果.结果表明,合金化元素的添加能够有效提高W在Cu液相中的溶解度,改善W和Cu的界面结合.Li等[11]在W-Cu体系中添加Ag元素,通过元素活化作用在较低烧结温度下获得了致密化良好的合金材料,且该体系具有良好的力学性能.Borji等[12]研究了合金化元素对W-Cu材料耐蚀性的影响,发现元素Ni可在促进W-Cu材料烧结致密化的同时增强体系的耐蚀性.Chookajorn等[13]在W体系中加入合金化元素Ti,制备了纳米晶W-Ti体系,其在1100 ℃的高温条件下仍能保持纳米晶晶粒组织,表明特定的合金化元素添加可显著增强纳米晶合金体系的热稳定性. ...
Microstructure characterization of W-Cu alloy sheets produced by high temperature and high pressure deformation technique
1
2017
... W-Cu材料由于其良好的力学性能和功能特性,在航空航天、电子信息、国防工业、电工和机械加工等领域[1,2,3,4,5,6]均有广泛运用.研究[7,8]表明,某些特定合金化元素添加可有效提高合金材料的使用性能和稳定性.例如,Chen等[9,10]研究了Fe、Co、Ni等元素对W-Cu粉末烧结过程的活化效果.结果表明,合金化元素的添加能够有效提高W在Cu液相中的溶解度,改善W和Cu的界面结合.Li等[11]在W-Cu体系中添加Ag元素,通过元素活化作用在较低烧结温度下获得了致密化良好的合金材料,且该体系具有良好的力学性能.Borji等[12]研究了合金化元素对W-Cu材料耐蚀性的影响,发现元素Ni可在促进W-Cu材料烧结致密化的同时增强体系的耐蚀性.Chookajorn等[13]在W体系中加入合金化元素Ti,制备了纳米晶W-Ti体系,其在1100 ℃的高温条件下仍能保持纳米晶晶粒组织,表明特定的合金化元素添加可显著增强纳米晶合金体系的热稳定性. ...
Tensile properties and deformation micromechanism of Ti-based metallic glass composite containing impurity elements
1
2019
... W-Cu材料由于其良好的力学性能和功能特性,在航空航天、电子信息、国防工业、电工和机械加工等领域[1,2,3,4,5,6]均有广泛运用.研究[7,8]表明,某些特定合金化元素添加可有效提高合金材料的使用性能和稳定性.例如,Chen等[9,10]研究了Fe、Co、Ni等元素对W-Cu粉末烧结过程的活化效果.结果表明,合金化元素的添加能够有效提高W在Cu液相中的溶解度,改善W和Cu的界面结合.Li等[11]在W-Cu体系中添加Ag元素,通过元素活化作用在较低烧结温度下获得了致密化良好的合金材料,且该体系具有良好的力学性能.Borji等[12]研究了合金化元素对W-Cu材料耐蚀性的影响,发现元素Ni可在促进W-Cu材料烧结致密化的同时增强体系的耐蚀性.Chookajorn等[13]在W体系中加入合金化元素Ti,制备了纳米晶W-Ti体系,其在1100 ℃的高温条件下仍能保持纳米晶晶粒组织,表明特定的合金化元素添加可显著增强纳米晶合金体系的热稳定性. ...
High-temperature grain size stabilization of nanocrystalline Fe-Cr alloys with Hf additions
1
2014
... W-Cu材料由于其良好的力学性能和功能特性,在航空航天、电子信息、国防工业、电工和机械加工等领域[1,2,3,4,5,6]均有广泛运用.研究[7,8]表明,某些特定合金化元素添加可有效提高合金材料的使用性能和稳定性.例如,Chen等[9,10]研究了Fe、Co、Ni等元素对W-Cu粉末烧结过程的活化效果.结果表明,合金化元素的添加能够有效提高W在Cu液相中的溶解度,改善W和Cu的界面结合.Li等[11]在W-Cu体系中添加Ag元素,通过元素活化作用在较低烧结温度下获得了致密化良好的合金材料,且该体系具有良好的力学性能.Borji等[12]研究了合金化元素对W-Cu材料耐蚀性的影响,发现元素Ni可在促进W-Cu材料烧结致密化的同时增强体系的耐蚀性.Chookajorn等[13]在W体系中加入合金化元素Ti,制备了纳米晶W-Ti体系,其在1100 ℃的高温条件下仍能保持纳米晶晶粒组织,表明特定的合金化元素添加可显著增强纳米晶合金体系的热稳定性. ...
The mechanical properties of W-Cu composite by activated sintering
1
2013
... W-Cu材料由于其良好的力学性能和功能特性,在航空航天、电子信息、国防工业、电工和机械加工等领域[1,2,3,4,5,6]均有广泛运用.研究[7,8]表明,某些特定合金化元素添加可有效提高合金材料的使用性能和稳定性.例如,Chen等[9,10]研究了Fe、Co、Ni等元素对W-Cu粉末烧结过程的活化效果.结果表明,合金化元素的添加能够有效提高W在Cu液相中的溶解度,改善W和Cu的界面结合.Li等[11]在W-Cu体系中添加Ag元素,通过元素活化作用在较低烧结温度下获得了致密化良好的合金材料,且该体系具有良好的力学性能.Borji等[12]研究了合金化元素对W-Cu材料耐蚀性的影响,发现元素Ni可在促进W-Cu材料烧结致密化的同时增强体系的耐蚀性.Chookajorn等[13]在W体系中加入合金化元素Ti,制备了纳米晶W-Ti体系,其在1100 ℃的高温条件下仍能保持纳米晶晶粒组织,表明特定的合金化元素添加可显著增强纳米晶合金体系的热稳定性. ...
Thermal and electrical properties of W-Cu composite produced by activated sintering
1
2013
... W-Cu材料由于其良好的力学性能和功能特性,在航空航天、电子信息、国防工业、电工和机械加工等领域[1,2,3,4,5,6]均有广泛运用.研究[7,8]表明,某些特定合金化元素添加可有效提高合金材料的使用性能和稳定性.例如,Chen等[9,10]研究了Fe、Co、Ni等元素对W-Cu粉末烧结过程的活化效果.结果表明,合金化元素的添加能够有效提高W在Cu液相中的溶解度,改善W和Cu的界面结合.Li等[11]在W-Cu体系中添加Ag元素,通过元素活化作用在较低烧结温度下获得了致密化良好的合金材料,且该体系具有良好的力学性能.Borji等[12]研究了合金化元素对W-Cu材料耐蚀性的影响,发现元素Ni可在促进W-Cu材料烧结致密化的同时增强体系的耐蚀性.Chookajorn等[13]在W体系中加入合金化元素Ti,制备了纳米晶W-Ti体系,其在1100 ℃的高温条件下仍能保持纳米晶晶粒组织,表明特定的合金化元素添加可显著增强纳米晶合金体系的热稳定性. ...
Densification and properties investigation of W-Cu composites prepared by electroless-plating and activated sintering
1
2018
... W-Cu材料由于其良好的力学性能和功能特性,在航空航天、电子信息、国防工业、电工和机械加工等领域[1,2,3,4,5,6]均有广泛运用.研究[7,8]表明,某些特定合金化元素添加可有效提高合金材料的使用性能和稳定性.例如,Chen等[9,10]研究了Fe、Co、Ni等元素对W-Cu粉末烧结过程的活化效果.结果表明,合金化元素的添加能够有效提高W在Cu液相中的溶解度,改善W和Cu的界面结合.Li等[11]在W-Cu体系中添加Ag元素,通过元素活化作用在较低烧结温度下获得了致密化良好的合金材料,且该体系具有良好的力学性能.Borji等[12]研究了合金化元素对W-Cu材料耐蚀性的影响,发现元素Ni可在促进W-Cu材料烧结致密化的同时增强体系的耐蚀性.Chookajorn等[13]在W体系中加入合金化元素Ti,制备了纳米晶W-Ti体系,其在1100 ℃的高温条件下仍能保持纳米晶晶粒组织,表明特定的合金化元素添加可显著增强纳米晶合金体系的热稳定性. ...
The effect of sintering activator on the erosion behavior of infiltrated W-10wt% Cu composite
1
2017
... W-Cu材料由于其良好的力学性能和功能特性,在航空航天、电子信息、国防工业、电工和机械加工等领域[1,2,3,4,5,6]均有广泛运用.研究[7,8]表明,某些特定合金化元素添加可有效提高合金材料的使用性能和稳定性.例如,Chen等[9,10]研究了Fe、Co、Ni等元素对W-Cu粉末烧结过程的活化效果.结果表明,合金化元素的添加能够有效提高W在Cu液相中的溶解度,改善W和Cu的界面结合.Li等[11]在W-Cu体系中添加Ag元素,通过元素活化作用在较低烧结温度下获得了致密化良好的合金材料,且该体系具有良好的力学性能.Borji等[12]研究了合金化元素对W-Cu材料耐蚀性的影响,发现元素Ni可在促进W-Cu材料烧结致密化的同时增强体系的耐蚀性.Chookajorn等[13]在W体系中加入合金化元素Ti,制备了纳米晶W-Ti体系,其在1100 ℃的高温条件下仍能保持纳米晶晶粒组织,表明特定的合金化元素添加可显著增强纳米晶合金体系的热稳定性. ...
Design of stable nanocrystalline alloys
1
2012
... W-Cu材料由于其良好的力学性能和功能特性,在航空航天、电子信息、国防工业、电工和机械加工等领域[1,2,3,4,5,6]均有广泛运用.研究[7,8]表明,某些特定合金化元素添加可有效提高合金材料的使用性能和稳定性.例如,Chen等[9,10]研究了Fe、Co、Ni等元素对W-Cu粉末烧结过程的活化效果.结果表明,合金化元素的添加能够有效提高W在Cu液相中的溶解度,改善W和Cu的界面结合.Li等[11]在W-Cu体系中添加Ag元素,通过元素活化作用在较低烧结温度下获得了致密化良好的合金材料,且该体系具有良好的力学性能.Borji等[12]研究了合金化元素对W-Cu材料耐蚀性的影响,发现元素Ni可在促进W-Cu材料烧结致密化的同时增强体系的耐蚀性.Chookajorn等[13]在W体系中加入合金化元素Ti,制备了纳米晶W-Ti体系,其在1100 ℃的高温条件下仍能保持纳米晶晶粒组织,表明特定的合金化元素添加可显著增强纳米晶合金体系的热稳定性. ...
How well can physical, chemical, and mechanical properties of materials be predicted by ab initio techniques?
2
2001
... 为了揭示各类合金化元素对合金体系的影响机制,有效进行合金化元素优选,研究人员进行了广泛的理论计算.各类方法中,经典热力学计算和Monte Carlo方法所需经验参数较多,分子动力学模拟受限于势函数,因此上述方法均较难分析合金化元素的微观作用机理,从而难以实现关于溶质元素属性预测和优选的目的.现有计算方法中,第一性原理计算作为微观尺度计算中的重要方法之一,能够对体系的晶体结构、键合作用、扩散过程和相变过程等进行量化描述,在W-Cu合金及其相关材料体系中得到了广泛应用[14].具体来看,对于W-Cu合金的元素掺杂体系,第一性原理可以从体系电子结构和能量的角度对界面结合强度、溶质扩散和偏聚等进行计算,且所需经验参数极少,物理意义明确[15,16].例如,Liang等[17]采用高通量第一性原理计算方法研究了W-Cu固溶体,对多种占比的W-Cu梯度界面进行了电子结构和成键方式的分析.Terakura等[18]获得了Cu-Ag和Cu-Au二元合金的相稳定性和体系能量的关系,第一性原理计算结果与相关实验结果一致.姜迪友[19]采用第一性原理方法研究了Y、Ti、Ta等合金化元素对钨合金体系的力学性能影响,讨论了模量、Poisson比和Cauchy应力等理化性质与材料性能之间的关联.另一方面,在第一性原理界面计算研究方面,Wu等[20]系统研究了钨基体系不同合金化元素影响下的溶质偏聚能和界面断裂功,计算了多类合金化元素对界面稳定性的影响趋势,揭示了合金化元素对钨基合金材料特征晶界的成键特性和力学性能影响机制.Zhou等[21]通过第一性原理拉伸模拟,分别计算了单晶W、纯W晶界和存在溶质偏聚情况的W晶界在拉伸断裂过程中的应力-应变曲线,系统分析了H原子偏析对于W晶界结合强度的影响机制.综上,基于第一性原理可有效探索不同合金化元素对W-Cu各类合金相和界面特征的作用机理. ...
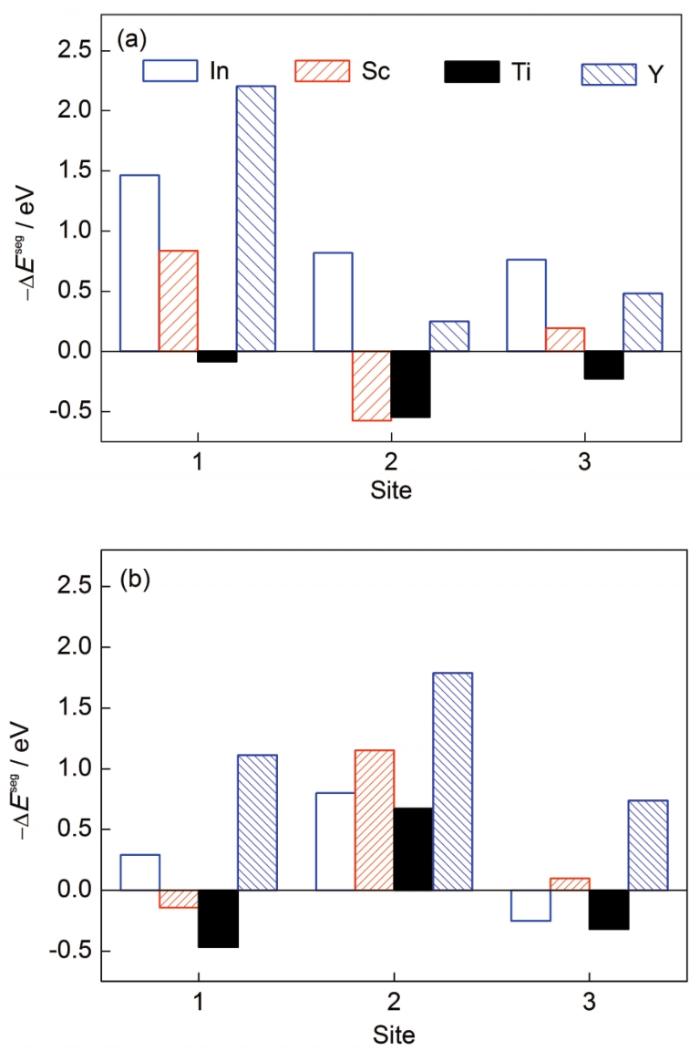
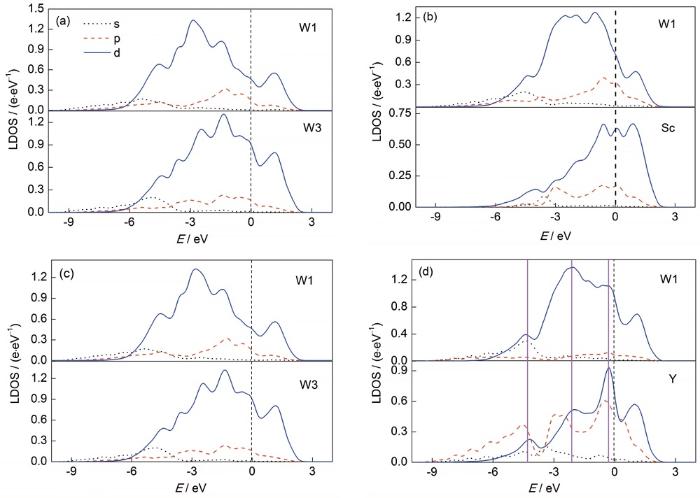
... 分别计算了W晶界和Cu晶界不同偏聚位点下的溶质偏聚能,如图4所示.可以发现,W晶界中位点1 (界面中心)为最优偏聚的位点,此时4类溶质元素均具有最低的溶质偏聚能,其中Sc、In和Y元素偏聚能低于-0.5 eV,符合强偏聚特征.根据已报道的实验结果[25],在W-Sc体系中,Sc元素的确具有极强的偏聚趋势,其在晶界处具有较高的溶质浓度,可有效提升W-Sc纳米晶体系的界面稳定性,这与模型预测吻合.在W-Ti体系中,Ti元素体现出较强的偏聚能力和较高的界面溶质浓度,与模型预测结果存在差异[14].这主要是由于实验采用了W-20%Ti (原子分数)体系,Ti的溶质浓度较高,而模型计算预测的是溶质元素在不考虑溶质浓度情况下的本征偏聚能力,因此存在一定的差异.Cu晶界中位点2 (界面松位)为最优偏聚位点,Sc、Ti、Y、In均具有强偏聚趋势,尤其是Y元素的偏聚能接近-2 eV,显示出Y原子在Cu基体中极强的晶界偏聚能力. ...
Grain boundary impurities in iron
1
2005
... 为了揭示各类合金化元素对合金体系的影响机制,有效进行合金化元素优选,研究人员进行了广泛的理论计算.各类方法中,经典热力学计算和Monte Carlo方法所需经验参数较多,分子动力学模拟受限于势函数,因此上述方法均较难分析合金化元素的微观作用机理,从而难以实现关于溶质元素属性预测和优选的目的.现有计算方法中,第一性原理计算作为微观尺度计算中的重要方法之一,能够对体系的晶体结构、键合作用、扩散过程和相变过程等进行量化描述,在W-Cu合金及其相关材料体系中得到了广泛应用[14].具体来看,对于W-Cu合金的元素掺杂体系,第一性原理可以从体系电子结构和能量的角度对界面结合强度、溶质扩散和偏聚等进行计算,且所需经验参数极少,物理意义明确[15,16].例如,Liang等[17]采用高通量第一性原理计算方法研究了W-Cu固溶体,对多种占比的W-Cu梯度界面进行了电子结构和成键方式的分析.Terakura等[18]获得了Cu-Ag和Cu-Au二元合金的相稳定性和体系能量的关系,第一性原理计算结果与相关实验结果一致.姜迪友[19]采用第一性原理方法研究了Y、Ti、Ta等合金化元素对钨合金体系的力学性能影响,讨论了模量、Poisson比和Cauchy应力等理化性质与材料性能之间的关联.另一方面,在第一性原理界面计算研究方面,Wu等[20]系统研究了钨基体系不同合金化元素影响下的溶质偏聚能和界面断裂功,计算了多类合金化元素对界面稳定性的影响趋势,揭示了合金化元素对钨基合金材料特征晶界的成键特性和力学性能影响机制.Zhou等[21]通过第一性原理拉伸模拟,分别计算了单晶W、纯W晶界和存在溶质偏聚情况的W晶界在拉伸断裂过程中的应力-应变曲线,系统分析了H原子偏析对于W晶界结合强度的影响机制.综上,基于第一性原理可有效探索不同合金化元素对W-Cu各类合金相和界面特征的作用机理. ...
Energetics of segregation and embrittling potency for non-transition elements in the Ni Σ5(012) symmetrical tilt grain boundary: A first-principles study
1
2004
... 为了揭示各类合金化元素对合金体系的影响机制,有效进行合金化元素优选,研究人员进行了广泛的理论计算.各类方法中,经典热力学计算和Monte Carlo方法所需经验参数较多,分子动力学模拟受限于势函数,因此上述方法均较难分析合金化元素的微观作用机理,从而难以实现关于溶质元素属性预测和优选的目的.现有计算方法中,第一性原理计算作为微观尺度计算中的重要方法之一,能够对体系的晶体结构、键合作用、扩散过程和相变过程等进行量化描述,在W-Cu合金及其相关材料体系中得到了广泛应用[14].具体来看,对于W-Cu合金的元素掺杂体系,第一性原理可以从体系电子结构和能量的角度对界面结合强度、溶质扩散和偏聚等进行计算,且所需经验参数极少,物理意义明确[15,16].例如,Liang等[17]采用高通量第一性原理计算方法研究了W-Cu固溶体,对多种占比的W-Cu梯度界面进行了电子结构和成键方式的分析.Terakura等[18]获得了Cu-Ag和Cu-Au二元合金的相稳定性和体系能量的关系,第一性原理计算结果与相关实验结果一致.姜迪友[19]采用第一性原理方法研究了Y、Ti、Ta等合金化元素对钨合金体系的力学性能影响,讨论了模量、Poisson比和Cauchy应力等理化性质与材料性能之间的关联.另一方面,在第一性原理界面计算研究方面,Wu等[20]系统研究了钨基体系不同合金化元素影响下的溶质偏聚能和界面断裂功,计算了多类合金化元素对界面稳定性的影响趋势,揭示了合金化元素对钨基合金材料特征晶界的成键特性和力学性能影响机制.Zhou等[21]通过第一性原理拉伸模拟,分别计算了单晶W、纯W晶界和存在溶质偏聚情况的W晶界在拉伸断裂过程中的应力-应变曲线,系统分析了H原子偏析对于W晶界结合强度的影响机制.综上,基于第一性原理可有效探索不同合金化元素对W-Cu各类合金相和界面特征的作用机理. ...
Cohesion strength and atomic structure of W-Cu graded interfaces
2
2017
... 为了揭示各类合金化元素对合金体系的影响机制,有效进行合金化元素优选,研究人员进行了广泛的理论计算.各类方法中,经典热力学计算和Monte Carlo方法所需经验参数较多,分子动力学模拟受限于势函数,因此上述方法均较难分析合金化元素的微观作用机理,从而难以实现关于溶质元素属性预测和优选的目的.现有计算方法中,第一性原理计算作为微观尺度计算中的重要方法之一,能够对体系的晶体结构、键合作用、扩散过程和相变过程等进行量化描述,在W-Cu合金及其相关材料体系中得到了广泛应用[14].具体来看,对于W-Cu合金的元素掺杂体系,第一性原理可以从体系电子结构和能量的角度对界面结合强度、溶质扩散和偏聚等进行计算,且所需经验参数极少,物理意义明确[15,16].例如,Liang等[17]采用高通量第一性原理计算方法研究了W-Cu固溶体,对多种占比的W-Cu梯度界面进行了电子结构和成键方式的分析.Terakura等[18]获得了Cu-Ag和Cu-Au二元合金的相稳定性和体系能量的关系,第一性原理计算结果与相关实验结果一致.姜迪友[19]采用第一性原理方法研究了Y、Ti、Ta等合金化元素对钨合金体系的力学性能影响,讨论了模量、Poisson比和Cauchy应力等理化性质与材料性能之间的关联.另一方面,在第一性原理界面计算研究方面,Wu等[20]系统研究了钨基体系不同合金化元素影响下的溶质偏聚能和界面断裂功,计算了多类合金化元素对界面稳定性的影响趋势,揭示了合金化元素对钨基合金材料特征晶界的成键特性和力学性能影响机制.Zhou等[21]通过第一性原理拉伸模拟,分别计算了单晶W、纯W晶界和存在溶质偏聚情况的W晶界在拉伸断裂过程中的应力-应变曲线,系统分析了H原子偏析对于W晶界结合强度的影响机制.综上,基于第一性原理可有效探索不同合金化元素对W-Cu各类合金相和界面特征的作用机理. ...
... 式中,Ω为W(110)/Cu(111)相界面面积(6.9 nm2),A1和A2分别为构建的W(110)和Cu(111)表面模型的表面积(W(110):7.2 nm2;Cu(111):5.7 nm2).根据式(1)可计算W(110)/Cu(111)相界错配度为-6.9%,综合考虑模型精度和计算成本,弹性应变对该模型的溶质偏聚计算无较大影响.实际上,W(110)/Cu(111)相界面在W-Cu体系的研究中也具有较高的相界面代表性,例如Jaouen等[36]研究了W(110)/Cu(111)多层材料的弹性应变和增强应力松弛效应;Liang等[17]利用第一性原理方法通过纯W与纯Cu界面计算确定了W(110)/Cu(111)界面为Cu沉积在W上的稳定界面. ...
Electronic theory of the alloy phase stability of Cu-Ag, Cu-Au, and Ag-Au systems
1
1987
... 为了揭示各类合金化元素对合金体系的影响机制,有效进行合金化元素优选,研究人员进行了广泛的理论计算.各类方法中,经典热力学计算和Monte Carlo方法所需经验参数较多,分子动力学模拟受限于势函数,因此上述方法均较难分析合金化元素的微观作用机理,从而难以实现关于溶质元素属性预测和优选的目的.现有计算方法中,第一性原理计算作为微观尺度计算中的重要方法之一,能够对体系的晶体结构、键合作用、扩散过程和相变过程等进行量化描述,在W-Cu合金及其相关材料体系中得到了广泛应用[14].具体来看,对于W-Cu合金的元素掺杂体系,第一性原理可以从体系电子结构和能量的角度对界面结合强度、溶质扩散和偏聚等进行计算,且所需经验参数极少,物理意义明确[15,16].例如,Liang等[17]采用高通量第一性原理计算方法研究了W-Cu固溶体,对多种占比的W-Cu梯度界面进行了电子结构和成键方式的分析.Terakura等[18]获得了Cu-Ag和Cu-Au二元合金的相稳定性和体系能量的关系,第一性原理计算结果与相关实验结果一致.姜迪友[19]采用第一性原理方法研究了Y、Ti、Ta等合金化元素对钨合金体系的力学性能影响,讨论了模量、Poisson比和Cauchy应力等理化性质与材料性能之间的关联.另一方面,在第一性原理界面计算研究方面,Wu等[20]系统研究了钨基体系不同合金化元素影响下的溶质偏聚能和界面断裂功,计算了多类合金化元素对界面稳定性的影响趋势,揭示了合金化元素对钨基合金材料特征晶界的成键特性和力学性能影响机制.Zhou等[21]通过第一性原理拉伸模拟,分别计算了单晶W、纯W晶界和存在溶质偏聚情况的W晶界在拉伸断裂过程中的应力-应变曲线,系统分析了H原子偏析对于W晶界结合强度的影响机制.综上,基于第一性原理可有效探索不同合金化元素对W-Cu各类合金相和界面特征的作用机理. ...
面向等离子体材料钨合金力学性质的第一性原理研究
1
2017
... 为了揭示各类合金化元素对合金体系的影响机制,有效进行合金化元素优选,研究人员进行了广泛的理论计算.各类方法中,经典热力学计算和Monte Carlo方法所需经验参数较多,分子动力学模拟受限于势函数,因此上述方法均较难分析合金化元素的微观作用机理,从而难以实现关于溶质元素属性预测和优选的目的.现有计算方法中,第一性原理计算作为微观尺度计算中的重要方法之一,能够对体系的晶体结构、键合作用、扩散过程和相变过程等进行量化描述,在W-Cu合金及其相关材料体系中得到了广泛应用[14].具体来看,对于W-Cu合金的元素掺杂体系,第一性原理可以从体系电子结构和能量的角度对界面结合强度、溶质扩散和偏聚等进行计算,且所需经验参数极少,物理意义明确[15,16].例如,Liang等[17]采用高通量第一性原理计算方法研究了W-Cu固溶体,对多种占比的W-Cu梯度界面进行了电子结构和成键方式的分析.Terakura等[18]获得了Cu-Ag和Cu-Au二元合金的相稳定性和体系能量的关系,第一性原理计算结果与相关实验结果一致.姜迪友[19]采用第一性原理方法研究了Y、Ti、Ta等合金化元素对钨合金体系的力学性能影响,讨论了模量、Poisson比和Cauchy应力等理化性质与材料性能之间的关联.另一方面,在第一性原理界面计算研究方面,Wu等[20]系统研究了钨基体系不同合金化元素影响下的溶质偏聚能和界面断裂功,计算了多类合金化元素对界面稳定性的影响趋势,揭示了合金化元素对钨基合金材料特征晶界的成键特性和力学性能影响机制.Zhou等[21]通过第一性原理拉伸模拟,分别计算了单晶W、纯W晶界和存在溶质偏聚情况的W晶界在拉伸断裂过程中的应力-应变曲线,系统分析了H原子偏析对于W晶界结合强度的影响机制.综上,基于第一性原理可有效探索不同合金化元素对W-Cu各类合金相和界面特征的作用机理. ...
面向等离子体材料钨合金力学性质的第一性原理研究
1
2017
... 为了揭示各类合金化元素对合金体系的影响机制,有效进行合金化元素优选,研究人员进行了广泛的理论计算.各类方法中,经典热力学计算和Monte Carlo方法所需经验参数较多,分子动力学模拟受限于势函数,因此上述方法均较难分析合金化元素的微观作用机理,从而难以实现关于溶质元素属性预测和优选的目的.现有计算方法中,第一性原理计算作为微观尺度计算中的重要方法之一,能够对体系的晶体结构、键合作用、扩散过程和相变过程等进行量化描述,在W-Cu合金及其相关材料体系中得到了广泛应用[14].具体来看,对于W-Cu合金的元素掺杂体系,第一性原理可以从体系电子结构和能量的角度对界面结合强度、溶质扩散和偏聚等进行计算,且所需经验参数极少,物理意义明确[15,16].例如,Liang等[17]采用高通量第一性原理计算方法研究了W-Cu固溶体,对多种占比的W-Cu梯度界面进行了电子结构和成键方式的分析.Terakura等[18]获得了Cu-Ag和Cu-Au二元合金的相稳定性和体系能量的关系,第一性原理计算结果与相关实验结果一致.姜迪友[19]采用第一性原理方法研究了Y、Ti、Ta等合金化元素对钨合金体系的力学性能影响,讨论了模量、Poisson比和Cauchy应力等理化性质与材料性能之间的关联.另一方面,在第一性原理界面计算研究方面,Wu等[20]系统研究了钨基体系不同合金化元素影响下的溶质偏聚能和界面断裂功,计算了多类合金化元素对界面稳定性的影响趋势,揭示了合金化元素对钨基合金材料特征晶界的成键特性和力学性能影响机制.Zhou等[21]通过第一性原理拉伸模拟,分别计算了单晶W、纯W晶界和存在溶质偏聚情况的W晶界在拉伸断裂过程中的应力-应变曲线,系统分析了H原子偏析对于W晶界结合强度的影响机制.综上,基于第一性原理可有效探索不同合金化元素对W-Cu各类合金相和界面特征的作用机理. ...
First-principles determination of grain boundary strengthening in tungsten: Dependence on grain boundary structure and metallic radius of solute
1
2016
... 为了揭示各类合金化元素对合金体系的影响机制,有效进行合金化元素优选,研究人员进行了广泛的理论计算.各类方法中,经典热力学计算和Monte Carlo方法所需经验参数较多,分子动力学模拟受限于势函数,因此上述方法均较难分析合金化元素的微观作用机理,从而难以实现关于溶质元素属性预测和优选的目的.现有计算方法中,第一性原理计算作为微观尺度计算中的重要方法之一,能够对体系的晶体结构、键合作用、扩散过程和相变过程等进行量化描述,在W-Cu合金及其相关材料体系中得到了广泛应用[14].具体来看,对于W-Cu合金的元素掺杂体系,第一性原理可以从体系电子结构和能量的角度对界面结合强度、溶质扩散和偏聚等进行计算,且所需经验参数极少,物理意义明确[15,16].例如,Liang等[17]采用高通量第一性原理计算方法研究了W-Cu固溶体,对多种占比的W-Cu梯度界面进行了电子结构和成键方式的分析.Terakura等[18]获得了Cu-Ag和Cu-Au二元合金的相稳定性和体系能量的关系,第一性原理计算结果与相关实验结果一致.姜迪友[19]采用第一性原理方法研究了Y、Ti、Ta等合金化元素对钨合金体系的力学性能影响,讨论了模量、Poisson比和Cauchy应力等理化性质与材料性能之间的关联.另一方面,在第一性原理界面计算研究方面,Wu等[20]系统研究了钨基体系不同合金化元素影响下的溶质偏聚能和界面断裂功,计算了多类合金化元素对界面稳定性的影响趋势,揭示了合金化元素对钨基合金材料特征晶界的成键特性和力学性能影响机制.Zhou等[21]通过第一性原理拉伸模拟,分别计算了单晶W、纯W晶界和存在溶质偏聚情况的W晶界在拉伸断裂过程中的应力-应变曲线,系统分析了H原子偏析对于W晶界结合强度的影响机制.综上,基于第一性原理可有效探索不同合金化元素对W-Cu各类合金相和界面特征的作用机理. ...
Effects of hydrogen on a tungsten grain boundary: A first-principles computational tensile test
1
2011
... 为了揭示各类合金化元素对合金体系的影响机制,有效进行合金化元素优选,研究人员进行了广泛的理论计算.各类方法中,经典热力学计算和Monte Carlo方法所需经验参数较多,分子动力学模拟受限于势函数,因此上述方法均较难分析合金化元素的微观作用机理,从而难以实现关于溶质元素属性预测和优选的目的.现有计算方法中,第一性原理计算作为微观尺度计算中的重要方法之一,能够对体系的晶体结构、键合作用、扩散过程和相变过程等进行量化描述,在W-Cu合金及其相关材料体系中得到了广泛应用[14].具体来看,对于W-Cu合金的元素掺杂体系,第一性原理可以从体系电子结构和能量的角度对界面结合强度、溶质扩散和偏聚等进行计算,且所需经验参数极少,物理意义明确[15,16].例如,Liang等[17]采用高通量第一性原理计算方法研究了W-Cu固溶体,对多种占比的W-Cu梯度界面进行了电子结构和成键方式的分析.Terakura等[18]获得了Cu-Ag和Cu-Au二元合金的相稳定性和体系能量的关系,第一性原理计算结果与相关实验结果一致.姜迪友[19]采用第一性原理方法研究了Y、Ti、Ta等合金化元素对钨合金体系的力学性能影响,讨论了模量、Poisson比和Cauchy应力等理化性质与材料性能之间的关联.另一方面,在第一性原理界面计算研究方面,Wu等[20]系统研究了钨基体系不同合金化元素影响下的溶质偏聚能和界面断裂功,计算了多类合金化元素对界面稳定性的影响趋势,揭示了合金化元素对钨基合金材料特征晶界的成键特性和力学性能影响机制.Zhou等[21]通过第一性原理拉伸模拟,分别计算了单晶W、纯W晶界和存在溶质偏聚情况的W晶界在拉伸断裂过程中的应力-应变曲线,系统分析了H原子偏析对于W晶界结合强度的影响机制.综上,基于第一性原理可有效探索不同合金化元素对W-Cu各类合金相和界面特征的作用机理. ...
Mutual relationship between material removal rate and W-W interfacial features during ultra-high temperature erosion of infiltrated W-10wt.% Cu composite
1
2018
... 值得注意的是,W-Cu体系为不互溶体系,在形成的双相材料中存在较复杂的界面关系,同时包含W/Cu相界面、W晶界面和Cu晶界面等界面类型,但目前的理论计算研究多集中于单一W晶界或Cu晶界体系,难以直接反映W-Cu体系中溶质原子与复杂界面环境的相互作用关系,尤其是多种合金化元素在W-Cu材料多类界面中的扩散、偏聚、析出等行为的影响机制研究较为匮乏,无法为实际材料设计提供充分的理论依据,因此,不互溶体系中的界面计算方法亟待建立.例如,Ahangarkani等[22]研究了Ni和Co元素分别对W/W界面和W/Cu界面的影响,发现Ni和Co元素的添加会降低W/W界面结合强度,但却能有效提升W/Cu界面结合强度.这说明即使是相同的合金化元素,在W-Cu体系各类界面中的影响作用也可能不同,仍需在现有的第一性原理计算方法上进一步考虑到多类界面和多种元素之间的相互作用关系,并以此获得能够考虑实际材料体系界面和物相复杂性的合金化元素优选策略.因此,虽然现有文献报道Sc、Ti、In、Y、Cr等多种元素在单独的W或Cu体系中具有晶界强偏聚趋势或促进烧结致密化等作用[23,24,25,26,27],但其在W-Cu不互溶双相合金中的研究和应用还鲜见报道. ...
Solute segregation and thermal stability of nanocrystalline solid solution systems
3
2019
... 值得注意的是,W-Cu体系为不互溶体系,在形成的双相材料中存在较复杂的界面关系,同时包含W/Cu相界面、W晶界面和Cu晶界面等界面类型,但目前的理论计算研究多集中于单一W晶界或Cu晶界体系,难以直接反映W-Cu体系中溶质原子与复杂界面环境的相互作用关系,尤其是多种合金化元素在W-Cu材料多类界面中的扩散、偏聚、析出等行为的影响机制研究较为匮乏,无法为实际材料设计提供充分的理论依据,因此,不互溶体系中的界面计算方法亟待建立.例如,Ahangarkani等[22]研究了Ni和Co元素分别对W/W界面和W/Cu界面的影响,发现Ni和Co元素的添加会降低W/W界面结合强度,但却能有效提升W/Cu界面结合强度.这说明即使是相同的合金化元素,在W-Cu体系各类界面中的影响作用也可能不同,仍需在现有的第一性原理计算方法上进一步考虑到多类界面和多种元素之间的相互作用关系,并以此获得能够考虑实际材料体系界面和物相复杂性的合金化元素优选策略.因此,虽然现有文献报道Sc、Ti、In、Y、Cr等多种元素在单独的W或Cu体系中具有晶界强偏聚趋势或促进烧结致密化等作用[23,24,25,26,27],但其在W-Cu不互溶双相合金中的研究和应用还鲜见报道. ...
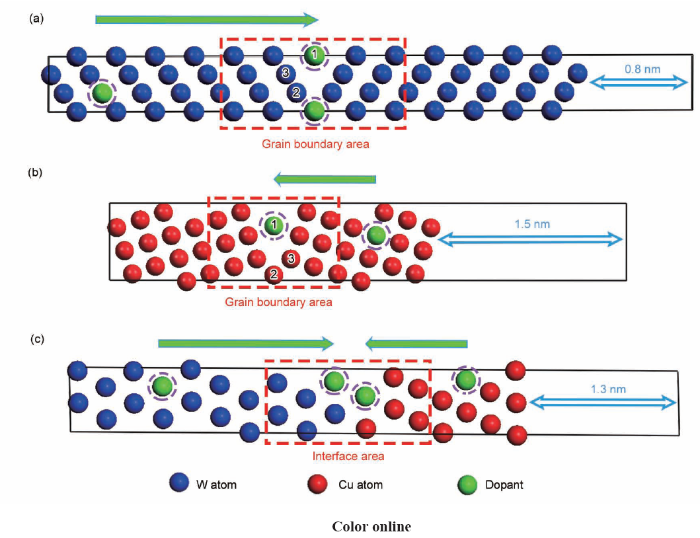
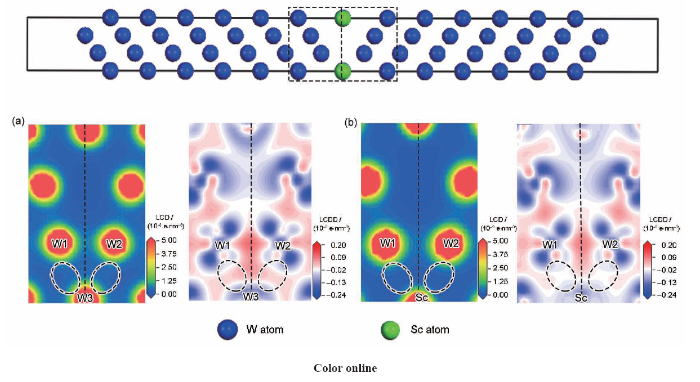
... 由于具有较高的晶界占比和较低的晶界能量,W体系中的3{111}<>晶界[23,28,29]和Cu体系中的5{310}<>晶界[30,31,32]具有较强的模型代表性和研究价值.在W晶界研究中,He等[33]基于3{111}<>界面采用第一性原理计算研究了V、Li、O、Ta等多种溶质原子的偏聚行为对晶界结构的影响作用;Tang等[23]基于3{111}<>晶界模型采用第一性原理与热力学计算相结合的方式系统研究了W-Sc偏聚体系的热稳定性;Scheiber等[29]则以3{111}<>界面为基础对多类过渡元素的表面偏聚和界面偏聚行为进行了系统研究.在Cu晶界研究中,王晓勉和秦湘阁[34]基于5{310}<>晶界采用分子动力学的方法研究了Cu纳米颗粒的烧结过程中的动力学影响机制,Tang等[31]以5{310}<>晶界模型为基础,采用第一性原理计算了Cu-Zn偏聚体系的偏聚能随溶质浓度的变化关系.本工作构建的W体系3{111}<>晶界模型和Cu体系5{310}<>晶界模型截面图如图1a和b所示,模型的真空层厚度分别0.8和1.5 nm,由于偏聚能定义为溶质偏聚前后晶界模型能量的差值,晶界模型两端包含的表面效应在溶质偏聚前后的2个界面模型能量差值计算中可以相互抵消,因此高指数表面及表面弛豫现象不会对模型精度造成较大影响. ...
... [23]基于3{111}<>晶界模型采用第一性原理与热力学计算相结合的方式系统研究了W-Sc偏聚体系的热稳定性;Scheiber等[29]则以3{111}<>界面为基础对多类过渡元素的表面偏聚和界面偏聚行为进行了系统研究.在Cu晶界研究中,王晓勉和秦湘阁[34]基于5{310}<>晶界采用分子动力学的方法研究了Cu纳米颗粒的烧结过程中的动力学影响机制,Tang等[31]以5{310}<>晶界模型为基础,采用第一性原理计算了Cu-Zn偏聚体系的偏聚能随溶质浓度的变化关系.本工作构建的W体系3{111}<>晶界模型和Cu体系5{310}<>晶界模型截面图如图1a和b所示,模型的真空层厚度分别0.8和1.5 nm,由于偏聚能定义为溶质偏聚前后晶界模型能量的差值,晶界模型两端包含的表面效应在溶质偏聚前后的2个界面模型能量差值计算中可以相互抵消,因此高指数表面及表面弛豫现象不会对模型精度造成较大影响. ...
Nanoscale segregation behavior and high-temperature stability of nanocrystalline W-20 at.% Ti
1
2014
... 值得注意的是,W-Cu体系为不互溶体系,在形成的双相材料中存在较复杂的界面关系,同时包含W/Cu相界面、W晶界面和Cu晶界面等界面类型,但目前的理论计算研究多集中于单一W晶界或Cu晶界体系,难以直接反映W-Cu体系中溶质原子与复杂界面环境的相互作用关系,尤其是多种合金化元素在W-Cu材料多类界面中的扩散、偏聚、析出等行为的影响机制研究较为匮乏,无法为实际材料设计提供充分的理论依据,因此,不互溶体系中的界面计算方法亟待建立.例如,Ahangarkani等[22]研究了Ni和Co元素分别对W/W界面和W/Cu界面的影响,发现Ni和Co元素的添加会降低W/W界面结合强度,但却能有效提升W/Cu界面结合强度.这说明即使是相同的合金化元素,在W-Cu体系各类界面中的影响作用也可能不同,仍需在现有的第一性原理计算方法上进一步考虑到多类界面和多种元素之间的相互作用关系,并以此获得能够考虑实际材料体系界面和物相复杂性的合金化元素优选策略.因此,虽然现有文献报道Sc、Ti、In、Y、Cr等多种元素在单独的W或Cu体系中具有晶界强偏聚趋势或促进烧结致密化等作用[23,24,25,26,27],但其在W-Cu不互溶双相合金中的研究和应用还鲜见报道. ...
Duplex nanocrystalline alloys: entropic nanostructure stabilization and a case study on W-Cr
2
2015
... 值得注意的是,W-Cu体系为不互溶体系,在形成的双相材料中存在较复杂的界面关系,同时包含W/Cu相界面、W晶界面和Cu晶界面等界面类型,但目前的理论计算研究多集中于单一W晶界或Cu晶界体系,难以直接反映W-Cu体系中溶质原子与复杂界面环境的相互作用关系,尤其是多种合金化元素在W-Cu材料多类界面中的扩散、偏聚、析出等行为的影响机制研究较为匮乏,无法为实际材料设计提供充分的理论依据,因此,不互溶体系中的界面计算方法亟待建立.例如,Ahangarkani等[22]研究了Ni和Co元素分别对W/W界面和W/Cu界面的影响,发现Ni和Co元素的添加会降低W/W界面结合强度,但却能有效提升W/Cu界面结合强度.这说明即使是相同的合金化元素,在W-Cu体系各类界面中的影响作用也可能不同,仍需在现有的第一性原理计算方法上进一步考虑到多类界面和多种元素之间的相互作用关系,并以此获得能够考虑实际材料体系界面和物相复杂性的合金化元素优选策略.因此,虽然现有文献报道Sc、Ti、In、Y、Cr等多种元素在单独的W或Cu体系中具有晶界强偏聚趋势或促进烧结致密化等作用[23,24,25,26,27],但其在W-Cu不互溶双相合金中的研究和应用还鲜见报道. ...
... 分别计算了W晶界和Cu晶界不同偏聚位点下的溶质偏聚能,如图4所示.可以发现,W晶界中位点1 (界面中心)为最优偏聚的位点,此时4类溶质元素均具有最低的溶质偏聚能,其中Sc、In和Y元素偏聚能低于-0.5 eV,符合强偏聚特征.根据已报道的实验结果[25],在W-Sc体系中,Sc元素的确具有极强的偏聚趋势,其在晶界处具有较高的溶质浓度,可有效提升W-Sc纳米晶体系的界面稳定性,这与模型预测吻合.在W-Ti体系中,Ti元素体现出较强的偏聚能力和较高的界面溶质浓度,与模型预测结果存在差异[14].这主要是由于实验采用了W-20%Ti (原子分数)体系,Ti的溶质浓度较高,而模型计算预测的是溶质元素在不考虑溶质浓度情况下的本征偏聚能力,因此存在一定的差异.Cu晶界中位点2 (界面松位)为最优偏聚位点,Sc、Ti、Y、In均具有强偏聚趋势,尤其是Y元素的偏聚能接近-2 eV,显示出Y原子在Cu基体中极强的晶界偏聚能力. ...
W-In体系溶质晶界偏聚行为的第一性原理计算
2
2019
... 值得注意的是,W-Cu体系为不互溶体系,在形成的双相材料中存在较复杂的界面关系,同时包含W/Cu相界面、W晶界面和Cu晶界面等界面类型,但目前的理论计算研究多集中于单一W晶界或Cu晶界体系,难以直接反映W-Cu体系中溶质原子与复杂界面环境的相互作用关系,尤其是多种合金化元素在W-Cu材料多类界面中的扩散、偏聚、析出等行为的影响机制研究较为匮乏,无法为实际材料设计提供充分的理论依据,因此,不互溶体系中的界面计算方法亟待建立.例如,Ahangarkani等[22]研究了Ni和Co元素分别对W/W界面和W/Cu界面的影响,发现Ni和Co元素的添加会降低W/W界面结合强度,但却能有效提升W/Cu界面结合强度.这说明即使是相同的合金化元素,在W-Cu体系各类界面中的影响作用也可能不同,仍需在现有的第一性原理计算方法上进一步考虑到多类界面和多种元素之间的相互作用关系,并以此获得能够考虑实际材料体系界面和物相复杂性的合金化元素优选策略.因此,虽然现有文献报道Sc、Ti、In、Y、Cr等多种元素在单独的W或Cu体系中具有晶界强偏聚趋势或促进烧结致密化等作用[23,24,25,26,27],但其在W-Cu不互溶双相合金中的研究和应用还鲜见报道. ...
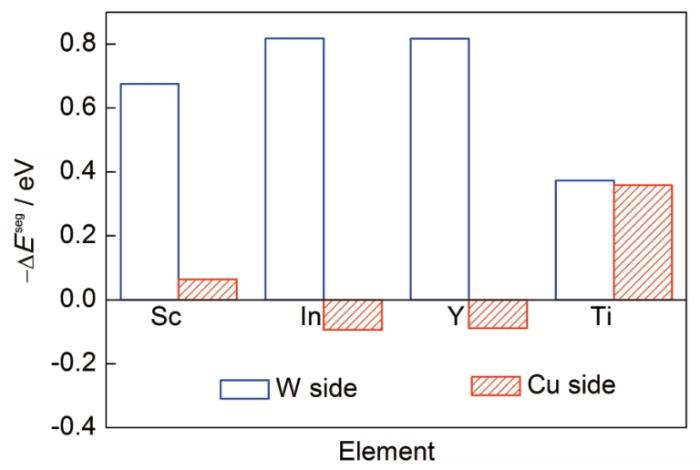
... 以Sc、Ti、Y、In 4种元素为例,计算溶质原子在W/Cu相界面的偏聚行为.溶质元素在相界面的偏聚分为W侧偏聚和Cu侧偏聚2种类型,计算结果如图3所示.可以看出,所计算的元素中Ti元素在W侧和Cu侧具有相近的偏聚能力,Sc、Y、In溶质元素在W侧的偏聚显著强于Cu侧的偏聚,且这3类元素在W侧的偏聚能显著低于-0.5 eV[26],体现出溶质在W/Cu相界面的强偏聚趋势. ...
W-In体系溶质晶界偏聚行为的第一性原理计算
2
2019
... 值得注意的是,W-Cu体系为不互溶体系,在形成的双相材料中存在较复杂的界面关系,同时包含W/Cu相界面、W晶界面和Cu晶界面等界面类型,但目前的理论计算研究多集中于单一W晶界或Cu晶界体系,难以直接反映W-Cu体系中溶质原子与复杂界面环境的相互作用关系,尤其是多种合金化元素在W-Cu材料多类界面中的扩散、偏聚、析出等行为的影响机制研究较为匮乏,无法为实际材料设计提供充分的理论依据,因此,不互溶体系中的界面计算方法亟待建立.例如,Ahangarkani等[22]研究了Ni和Co元素分别对W/W界面和W/Cu界面的影响,发现Ni和Co元素的添加会降低W/W界面结合强度,但却能有效提升W/Cu界面结合强度.这说明即使是相同的合金化元素,在W-Cu体系各类界面中的影响作用也可能不同,仍需在现有的第一性原理计算方法上进一步考虑到多类界面和多种元素之间的相互作用关系,并以此获得能够考虑实际材料体系界面和物相复杂性的合金化元素优选策略.因此,虽然现有文献报道Sc、Ti、In、Y、Cr等多种元素在单独的W或Cu体系中具有晶界强偏聚趋势或促进烧结致密化等作用[23,24,25,26,27],但其在W-Cu不互溶双相合金中的研究和应用还鲜见报道. ...
... 以Sc、Ti、Y、In 4种元素为例,计算溶质原子在W/Cu相界面的偏聚行为.溶质元素在相界面的偏聚分为W侧偏聚和Cu侧偏聚2种类型,计算结果如图3所示.可以看出,所计算的元素中Ti元素在W侧和Cu侧具有相近的偏聚能力,Sc、Y、In溶质元素在W侧的偏聚显著强于Cu侧的偏聚,且这3类元素在W侧的偏聚能显著低于-0.5 eV[26],体现出溶质在W/Cu相界面的强偏聚趋势. ...
First-principles investigation of structural, mechanical and electronic properties for Cu-Ti intermetallics
1
2016
... 值得注意的是,W-Cu体系为不互溶体系,在形成的双相材料中存在较复杂的界面关系,同时包含W/Cu相界面、W晶界面和Cu晶界面等界面类型,但目前的理论计算研究多集中于单一W晶界或Cu晶界体系,难以直接反映W-Cu体系中溶质原子与复杂界面环境的相互作用关系,尤其是多种合金化元素在W-Cu材料多类界面中的扩散、偏聚、析出等行为的影响机制研究较为匮乏,无法为实际材料设计提供充分的理论依据,因此,不互溶体系中的界面计算方法亟待建立.例如,Ahangarkani等[22]研究了Ni和Co元素分别对W/W界面和W/Cu界面的影响,发现Ni和Co元素的添加会降低W/W界面结合强度,但却能有效提升W/Cu界面结合强度.这说明即使是相同的合金化元素,在W-Cu体系各类界面中的影响作用也可能不同,仍需在现有的第一性原理计算方法上进一步考虑到多类界面和多种元素之间的相互作用关系,并以此获得能够考虑实际材料体系界面和物相复杂性的合金化元素优选策略.因此,虽然现有文献报道Sc、Ti、In、Y、Cr等多种元素在单独的W或Cu体系中具有晶界强偏聚趋势或促进烧结致密化等作用[23,24,25,26,27],但其在W-Cu不互溶双相合金中的研究和应用还鲜见报道. ...
Ab initio search for cohesion-enhancing solute elements at grain boundaries in molybdenum and tungsten
1
2016
... 由于具有较高的晶界占比和较低的晶界能量,W体系中的3{111}<>晶界[23,28,29]和Cu体系中的5{310}<>晶界[30,31,32]具有较强的模型代表性和研究价值.在W晶界研究中,He等[33]基于3{111}<>界面采用第一性原理计算研究了V、Li、O、Ta等多种溶质原子的偏聚行为对晶界结构的影响作用;Tang等[23]基于3{111}<>晶界模型采用第一性原理与热力学计算相结合的方式系统研究了W-Sc偏聚体系的热稳定性;Scheiber等[29]则以3{111}<>界面为基础对多类过渡元素的表面偏聚和界面偏聚行为进行了系统研究.在Cu晶界研究中,王晓勉和秦湘阁[34]基于5{310}<>晶界采用分子动力学的方法研究了Cu纳米颗粒的烧结过程中的动力学影响机制,Tang等[31]以5{310}<>晶界模型为基础,采用第一性原理计算了Cu-Zn偏聚体系的偏聚能随溶质浓度的变化关系.本工作构建的W体系3{111}<>晶界模型和Cu体系5{310}<>晶界模型截面图如图1a和b所示,模型的真空层厚度分别0.8和1.5 nm,由于偏聚能定义为溶质偏聚前后晶界模型能量的差值,晶界模型两端包含的表面效应在溶质偏聚前后的2个界面模型能量差值计算中可以相互抵消,因此高指数表面及表面弛豫现象不会对模型精度造成较大影响. ...
Ab-initio search for cohesion-enhancing solute elements at grain boundaries in molybdenum and tungsten
2
2016
... 由于具有较高的晶界占比和较低的晶界能量,W体系中的3{111}<>晶界[23,28,29]和Cu体系中的5{310}<>晶界[30,31,32]具有较强的模型代表性和研究价值.在W晶界研究中,He等[33]基于3{111}<>界面采用第一性原理计算研究了V、Li、O、Ta等多种溶质原子的偏聚行为对晶界结构的影响作用;Tang等[23]基于3{111}<>晶界模型采用第一性原理与热力学计算相结合的方式系统研究了W-Sc偏聚体系的热稳定性;Scheiber等[29]则以3{111}<>界面为基础对多类过渡元素的表面偏聚和界面偏聚行为进行了系统研究.在Cu晶界研究中,王晓勉和秦湘阁[34]基于5{310}<>晶界采用分子动力学的方法研究了Cu纳米颗粒的烧结过程中的动力学影响机制,Tang等[31]以5{310}<>晶界模型为基础,采用第一性原理计算了Cu-Zn偏聚体系的偏聚能随溶质浓度的变化关系.本工作构建的W体系3{111}<>晶界模型和Cu体系5{310}<>晶界模型截面图如图1a和b所示,模型的真空层厚度分别0.8和1.5 nm,由于偏聚能定义为溶质偏聚前后晶界模型能量的差值,晶界模型两端包含的表面效应在溶质偏聚前后的2个界面模型能量差值计算中可以相互抵消,因此高指数表面及表面弛豫现象不会对模型精度造成较大影响. ...
... [29]则以3{111}<>界面为基础对多类过渡元素的表面偏聚和界面偏聚行为进行了系统研究.在Cu晶界研究中,王晓勉和秦湘阁[34]基于5{310}<>晶界采用分子动力学的方法研究了Cu纳米颗粒的烧结过程中的动力学影响机制,Tang等[31]以5{310}<>晶界模型为基础,采用第一性原理计算了Cu-Zn偏聚体系的偏聚能随溶质浓度的变化关系.本工作构建的W体系3{111}<>晶界模型和Cu体系5{310}<>晶界模型截面图如图1a和b所示,模型的真空层厚度分别0.8和1.5 nm,由于偏聚能定义为溶质偏聚前后晶界模型能量的差值,晶界模型两端包含的表面效应在溶质偏聚前后的2个界面模型能量差值计算中可以相互抵消,因此高指数表面及表面弛豫现象不会对模型精度造成较大影响. ...
Advance in orientation microscopy: Quantitative analysis of nanocrystalline structures
1
2011
... 由于具有较高的晶界占比和较低的晶界能量,W体系中的3{111}<>晶界[23,28,29]和Cu体系中的5{310}<>晶界[30,31,32]具有较强的模型代表性和研究价值.在W晶界研究中,He等[33]基于3{111}<>界面采用第一性原理计算研究了V、Li、O、Ta等多种溶质原子的偏聚行为对晶界结构的影响作用;Tang等[23]基于3{111}<>晶界模型采用第一性原理与热力学计算相结合的方式系统研究了W-Sc偏聚体系的热稳定性;Scheiber等[29]则以3{111}<>界面为基础对多类过渡元素的表面偏聚和界面偏聚行为进行了系统研究.在Cu晶界研究中,王晓勉和秦湘阁[34]基于5{310}<>晶界采用分子动力学的方法研究了Cu纳米颗粒的烧结过程中的动力学影响机制,Tang等[31]以5{310}<>晶界模型为基础,采用第一性原理计算了Cu-Zn偏聚体系的偏聚能随溶质浓度的变化关系.本工作构建的W体系3{111}<>晶界模型和Cu体系5{310}<>晶界模型截面图如图1a和b所示,模型的真空层厚度分别0.8和1.5 nm,由于偏聚能定义为溶质偏聚前后晶界模型能量的差值,晶界模型两端包含的表面效应在溶质偏聚前后的2个界面模型能量差值计算中可以相互抵消,因此高指数表面及表面弛豫现象不会对模型精度造成较大影响. ...
Modeling of Li diffusion in nanocrystalline Li-Si anode material
2
2018
... 由于具有较高的晶界占比和较低的晶界能量,W体系中的3{111}<>晶界[23,28,29]和Cu体系中的5{310}<>晶界[30,31,32]具有较强的模型代表性和研究价值.在W晶界研究中,He等[33]基于3{111}<>界面采用第一性原理计算研究了V、Li、O、Ta等多种溶质原子的偏聚行为对晶界结构的影响作用;Tang等[23]基于3{111}<>晶界模型采用第一性原理与热力学计算相结合的方式系统研究了W-Sc偏聚体系的热稳定性;Scheiber等[29]则以3{111}<>界面为基础对多类过渡元素的表面偏聚和界面偏聚行为进行了系统研究.在Cu晶界研究中,王晓勉和秦湘阁[34]基于5{310}<>晶界采用分子动力学的方法研究了Cu纳米颗粒的烧结过程中的动力学影响机制,Tang等[31]以5{310}<>晶界模型为基础,采用第一性原理计算了Cu-Zn偏聚体系的偏聚能随溶质浓度的变化关系.本工作构建的W体系3{111}<>晶界模型和Cu体系5{310}<>晶界模型截面图如图1a和b所示,模型的真空层厚度分别0.8和1.5 nm,由于偏聚能定义为溶质偏聚前后晶界模型能量的差值,晶界模型两端包含的表面效应在溶质偏聚前后的2个界面模型能量差值计算中可以相互抵消,因此高指数表面及表面弛豫现象不会对模型精度造成较大影响. ...
... [31]以5{310}<>晶界模型为基础,采用第一性原理计算了Cu-Zn偏聚体系的偏聚能随溶质浓度的变化关系.本工作构建的W体系3{111}<>晶界模型和Cu体系5{310}<>晶界模型截面图如图1a和b所示,模型的真空层厚度分别0.8和1.5 nm,由于偏聚能定义为溶质偏聚前后晶界模型能量的差值,晶界模型两端包含的表面效应在溶质偏聚前后的2个界面模型能量差值计算中可以相互抵消,因此高指数表面及表面弛豫现象不会对模型精度造成较大影响. ...
Secondary recrystallization in copper
1
1949
... 由于具有较高的晶界占比和较低的晶界能量,W体系中的3{111}<>晶界[23,28,29]和Cu体系中的5{310}<>晶界[30,31,32]具有较强的模型代表性和研究价值.在W晶界研究中,He等[33]基于3{111}<>界面采用第一性原理计算研究了V、Li、O、Ta等多种溶质原子的偏聚行为对晶界结构的影响作用;Tang等[23]基于3{111}<>晶界模型采用第一性原理与热力学计算相结合的方式系统研究了W-Sc偏聚体系的热稳定性;Scheiber等[29]则以3{111}<>界面为基础对多类过渡元素的表面偏聚和界面偏聚行为进行了系统研究.在Cu晶界研究中,王晓勉和秦湘阁[34]基于5{310}<>晶界采用分子动力学的方法研究了Cu纳米颗粒的烧结过程中的动力学影响机制,Tang等[31]以5{310}<>晶界模型为基础,采用第一性原理计算了Cu-Zn偏聚体系的偏聚能随溶质浓度的变化关系.本工作构建的W体系3{111}<>晶界模型和Cu体系5{310}<>晶界模型截面图如图1a和b所示,模型的真空层厚度分别0.8和1.5 nm,由于偏聚能定义为溶质偏聚前后晶界模型能量的差值,晶界模型两端包含的表面效应在溶质偏聚前后的2个界面模型能量差值计算中可以相互抵消,因此高指数表面及表面弛豫现象不会对模型精度造成较大影响. ...
First-principles investigation of vacancies in LiTaO3
1
2016
... 由于具有较高的晶界占比和较低的晶界能量,W体系中的3{111}<>晶界[23,28,29]和Cu体系中的5{310}<>晶界[30,31,32]具有较强的模型代表性和研究价值.在W晶界研究中,He等[33]基于3{111}<>界面采用第一性原理计算研究了V、Li、O、Ta等多种溶质原子的偏聚行为对晶界结构的影响作用;Tang等[23]基于3{111}<>晶界模型采用第一性原理与热力学计算相结合的方式系统研究了W-Sc偏聚体系的热稳定性;Scheiber等[29]则以3{111}<>界面为基础对多类过渡元素的表面偏聚和界面偏聚行为进行了系统研究.在Cu晶界研究中,王晓勉和秦湘阁[34]基于5{310}<>晶界采用分子动力学的方法研究了Cu纳米颗粒的烧结过程中的动力学影响机制,Tang等[31]以5{310}<>晶界模型为基础,采用第一性原理计算了Cu-Zn偏聚体系的偏聚能随溶质浓度的变化关系.本工作构建的W体系3{111}<>晶界模型和Cu体系5{310}<>晶界模型截面图如图1a和b所示,模型的真空层厚度分别0.8和1.5 nm,由于偏聚能定义为溶质偏聚前后晶界模型能量的差值,晶界模型两端包含的表面效应在溶质偏聚前后的2个界面模型能量差值计算中可以相互抵消,因此高指数表面及表面弛豫现象不会对模型精度造成较大影响. ...
5晶界对铜纳米颗粒烧结行为的影响
1
2016
... 由于具有较高的晶界占比和较低的晶界能量,W体系中的3{111}<>晶界[23,28,29]和Cu体系中的5{310}<>晶界[30,31,32]具有较强的模型代表性和研究价值.在W晶界研究中,He等[33]基于3{111}<>界面采用第一性原理计算研究了V、Li、O、Ta等多种溶质原子的偏聚行为对晶界结构的影响作用;Tang等[23]基于3{111}<>晶界模型采用第一性原理与热力学计算相结合的方式系统研究了W-Sc偏聚体系的热稳定性;Scheiber等[29]则以3{111}<>界面为基础对多类过渡元素的表面偏聚和界面偏聚行为进行了系统研究.在Cu晶界研究中,王晓勉和秦湘阁[34]基于5{310}<>晶界采用分子动力学的方法研究了Cu纳米颗粒的烧结过程中的动力学影响机制,Tang等[31]以5{310}<>晶界模型为基础,采用第一性原理计算了Cu-Zn偏聚体系的偏聚能随溶质浓度的变化关系.本工作构建的W体系3{111}<>晶界模型和Cu体系5{310}<>晶界模型截面图如图1a和b所示,模型的真空层厚度分别0.8和1.5 nm,由于偏聚能定义为溶质偏聚前后晶界模型能量的差值,晶界模型两端包含的表面效应在溶质偏聚前后的2个界面模型能量差值计算中可以相互抵消,因此高指数表面及表面弛豫现象不会对模型精度造成较大影响. ...
5晶界对铜纳米颗粒烧结行为的影响
1
2016
... 由于具有较高的晶界占比和较低的晶界能量,W体系中的3{111}<>晶界[23,28,29]和Cu体系中的5{310}<>晶界[30,31,32]具有较强的模型代表性和研究价值.在W晶界研究中,He等[33]基于3{111}<>界面采用第一性原理计算研究了V、Li、O、Ta等多种溶质原子的偏聚行为对晶界结构的影响作用;Tang等[23]基于3{111}<>晶界模型采用第一性原理与热力学计算相结合的方式系统研究了W-Sc偏聚体系的热稳定性;Scheiber等[29]则以3{111}<>界面为基础对多类过渡元素的表面偏聚和界面偏聚行为进行了系统研究.在Cu晶界研究中,王晓勉和秦湘阁[34]基于5{310}<>晶界采用分子动力学的方法研究了Cu纳米颗粒的烧结过程中的动力学影响机制,Tang等[31]以5{310}<>晶界模型为基础,采用第一性原理计算了Cu-Zn偏聚体系的偏聚能随溶质浓度的变化关系.本工作构建的W体系3{111}<>晶界模型和Cu体系5{310}<>晶界模型截面图如图1a和b所示,模型的真空层厚度分别0.8和1.5 nm,由于偏聚能定义为溶质偏聚前后晶界模型能量的差值,晶界模型两端包含的表面效应在溶质偏聚前后的2个界面模型能量差值计算中可以相互抵消,因此高指数表面及表面弛豫现象不会对模型精度造成较大影响. ...
Adhesion at cerium doped metal-ceramic α-Fe/WC interface: A first-principles calculation
1
2019
... 另一方面,对于W/Cu相界面计算,则分别以W和Cu的(100)、(110)和(111)等低指数晶面为基础构建了W(100)/Cu(100)、W(110)/Cu(111)和W(111)/Cu(111)等多类相界面模型,并通过电子结构分析选取了W(110)/Cu(111)相界面进行溶质偏聚模拟.W(110)/Cu(111)相界模型如图1c所示,真空层厚度为1.3 nm且进行两端原子层固定,相界模型接触面分别为W(110)和Cu(111),晶带轴W[]//Cu[].根据界面错配度()公式[35]: ...
Elastic strains and enhanced stress relaxation effects induced by ion irradiation in W(110)/Cu(111) multilayers: Comparative EXAFS and X-ray diffraction studies
1
2001
... 式中,Ω为W(110)/Cu(111)相界面面积(6.9 nm2),A1和A2分别为构建的W(110)和Cu(111)表面模型的表面积(W(110):7.2 nm2;Cu(111):5.7 nm2).根据式(1)可计算W(110)/Cu(111)相界错配度为-6.9%,综合考虑模型精度和计算成本,弹性应变对该模型的溶质偏聚计算无较大影响.实际上,W(110)/Cu(111)相界面在W-Cu体系的研究中也具有较高的相界面代表性,例如Jaouen等[36]研究了W(110)/Cu(111)多层材料的弹性应变和增强应力松弛效应;Liang等[17]利用第一性原理方法通过纯W与纯Cu界面计算确定了W(110)/Cu(111)界面为Cu沉积在W上的稳定界面. ...
First-principles simulation: Ideas, illustrations and the CASTEP code
1
2002
... 本工作所有的计算都采用Materials Studio软件中的CASTEP[37]模块完成.计算中选择超软赝势(ultrasoft pseudopotential,Usp)[38],W和Cu的价电子结构分别为5d46s2和3d104s1,4种溶质原子Sc、Ti、Y、In的价电子结构分别为3d14s2、3d24s2、4d15s2和4d105s25p1.泛函形式采取GGA-PBE (Perdew-Burke-Ernzerhof generalized gradient approximation)方法[39].平面波截断能取400 eV,k点设置为8×8×1.各项收敛参数为能量小于2×10-6 eV/atom,原子间的相互作用力小于0.5 eV/nm,应力小于0.1 GPa,原子的最大位移变化小于0.0002 nm. ...
Soft self-consistent pseudopotentials in a generalized eigenvalue formalism
1
1990
... 本工作所有的计算都采用Materials Studio软件中的CASTEP[37]模块完成.计算中选择超软赝势(ultrasoft pseudopotential,Usp)[38],W和Cu的价电子结构分别为5d46s2和3d104s1,4种溶质原子Sc、Ti、Y、In的价电子结构分别为3d14s2、3d24s2、4d15s2和4d105s25p1.泛函形式采取GGA-PBE (Perdew-Burke-Ernzerhof generalized gradient approximation)方法[39].平面波截断能取400 eV,k点设置为8×8×1.各项收敛参数为能量小于2×10-6 eV/atom,原子间的相互作用力小于0.5 eV/nm,应力小于0.1 GPa,原子的最大位移变化小于0.0002 nm. ...
Generalized gradient approximation made simple
1
1996
... 本工作所有的计算都采用Materials Studio软件中的CASTEP[37]模块完成.计算中选择超软赝势(ultrasoft pseudopotential,Usp)[38],W和Cu的价电子结构分别为5d46s2和3d104s1,4种溶质原子Sc、Ti、Y、In的价电子结构分别为3d14s2、3d24s2、4d15s2和4d105s25p1.泛函形式采取GGA-PBE (Perdew-Burke-Ernzerhof generalized gradient approximation)方法[39].平面波截断能取400 eV,k点设置为8×8×1.各项收敛参数为能量小于2×10-6 eV/atom,原子间的相互作用力小于0.5 eV/nm,应力小于0.1 GPa,原子的最大位移变化小于0.0002 nm. ...
Segregation of alloying atoms at a tilt symmetric grain boundary in tungsten and their strengthening and embrittling effects
1
2014
... 本工作通过计算溶质原子从晶界较远处偏聚到晶界区域的能量变化获得溶质的偏聚能[40].偏聚能()计算公式如下[41]: ...
Ab initio description of segregation and cohesion of grain boundaries in W-25 at.% Re alloys
1
2015
... 本工作通过计算溶质原子从晶界较远处偏聚到晶界区域的能量变化获得溶质的偏聚能[40].偏聚能()计算公式如下[41]: ...
Hydrogen in aluminum: First-principles calculations of structure and thermodynamics
1
2004
... 式中,是偏聚后体系的总能量,是偏聚前体系的总能量.二者差值即为溶质原子偏聚过程引起的能量变化.偏聚能数值越负代表溶质原子的偏聚能力越强,利于提升界面稳定性.此外,为研究合金化元素与W-Cu基体的结合能力,分别在W体相和Cu体相中进行了形成能()的计算,计算公式如下[42]: ...






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}