Advanced Lubrication and Sealing Materials Research Center, State Key Laboratory of Solidification Technology, Northwestern Polytechnical University, Xi'an 710072, China
Recently, the rapid advancement of extreme non-equilibrium material processing and fabrication techniques, such as 3D printing and rapid die-casting, has led to the continuous development of new materials with exceptional properties. However, current non-equilibrium processing technologies face technical challenges, such as the lack of clear guidelines for process optimization, which considerably limits the advancement and application of advanced materials. The solidification and solid phase transformations involved in materials prepared through non-equilibrium processing pertain to a non-equilibrium dissipative system and manifest throughout the entire dynamic process of material fabrication. By investigating key scientific issues such as non-equilibrium phase transformation dynamics, non-equilibrium solute diffusion, and solute-drag effects, developing a theoretical framework for the entire non-equilibrium material processing, from solidification to solid phase transformation is possible. This not only provides theoretical support for the design and fabrication of non-equilibrium materials but also introduces novel concepts for optimizing process parameters in non-equilibrium processing technologies. This review is crucial for advancing non-equilibrium phase transformation theory and deepening our understanding of fundamental theoretical research. Interfaces play a critical role in microstructure control during material processing, thereby making an accurate theoretical description of their kinetics is especially important. This review focuses on the common characteristics of liquid/solid interfaces during melting, solid/liquid interfaces during solidification, and solid/solid interfaces during solid state phase transformations and summarizes and analyzes the history and current state of sharp-interface models for interface kinetics. Using the solidification of binary alloys as an example, the review first introduces interface kinetic theories under local non-equilibrium conditions, covering descriptions of interface kinetic processes and interface kinetic models for steady-state and non-steady-state conditions. The physical nature of one-step and two-step trans-interface diffusion is demonstrated. Next, the review describes interface kinetic theories under full non-equilibrium conditions by comparing the applications of the kinetic energy method and the effective mobility method for non-equilibrium solute diffusion in bulk phases. Thereafter, it introduces interface kinetic theories incorporating the partial solute drag effect present and discusses limitations in current methods for addressing partial solute drag. This study aims to enhance understanding of interface kinetics, offering insights into microstructure control. Finally, an outlook on the future of non-equilibrium interface kinetic theories is provided, which outlines directions for future research.
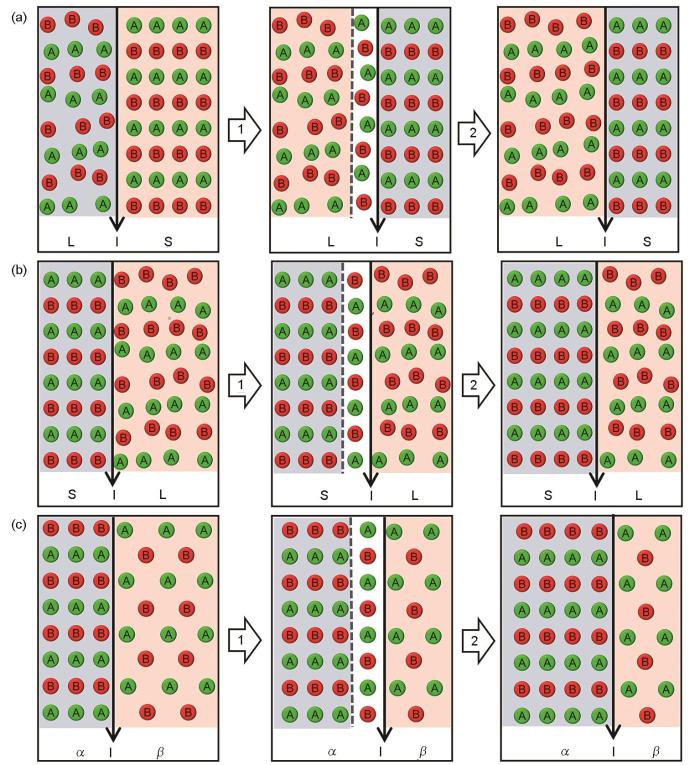
Fig.1
Schematics for the interface kinetic processes in different phase transformation types (L—liquid, S—solid, I—interface, A—solvent atom A, B—solute atom B, α—solid phase α, β—solid phase β, the same below)
(a) liquid/solid interface during melting
(b) solid/liquid interface during solidification
(c) solid/solid (α/β) interface during solid-state phase-transformation
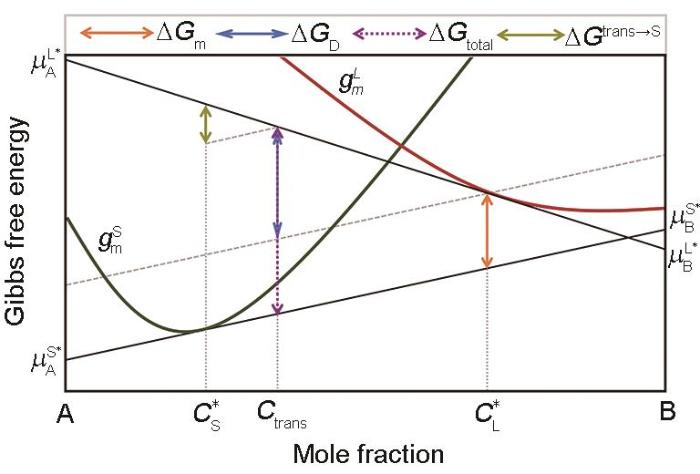
Fig.3
Mole Gibbs free energy diagram for the solid/liquid interface kinetic processes under a steady-state condition (Green and red curves represent the Gibbs free energy curve of the solid phase and liquid phase, respectively, the same below. Total Gibbs free energy dissipated by the interface after solidification of 1 mol liquid is = . By translating the tangent of Gibbs free energy curve of solid at to the Gibbs free energy curve of liquid at , is divided into two parts: The upper one for trans-interface diffusion is = and latter part for interface migration is = .—Gibbs free energy of interface migration, —Gibbs free energy of trans-interface diffusion, —the solid composition at the interface, —the liquid composition at the interface, —the chemical potential of A across the interface, —the chemical potential of B across the interface, and —the chemical potential at the interface for liquid and solid phase B, and —the chemical potential at the interface for liquid and solid phase B, respectively, —the mole Gibbs free energy of the solid, —the mole Gibbs free energy of the liquid)
Fig.4
Mole Gibbs free energy diagram for the solid/liquid interface kinetic processes under a non-steady-state condition (Total Gibbs free energy dissipated by the interface after solidification of 1 mol liquid is = . By translating the tangent of Gibbs free energy curve of solid at to the Gibbs free energy curve of liquid at , Gtotal is divided into two parts: The upper one for trans-interface diffusion is = and latter part for interface migration is = . The difference in Gtotal between the steady-state condition and the non-steady-state condition is = , which is the Gibbs free energy dissipated to adjust the actual composition transferred across the interface from to Ctrans (—the solute component that finally crosses the interface and enters the solid phase)
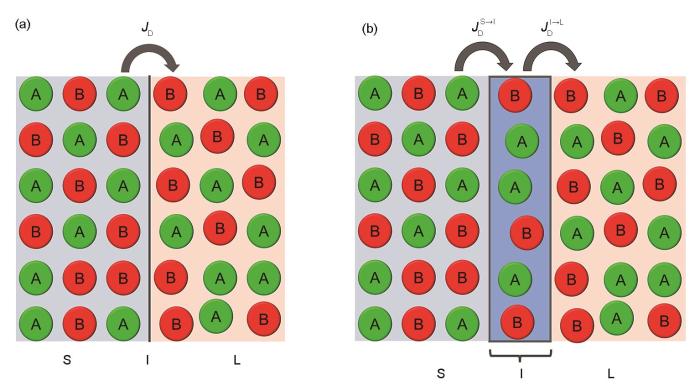
Fig.5
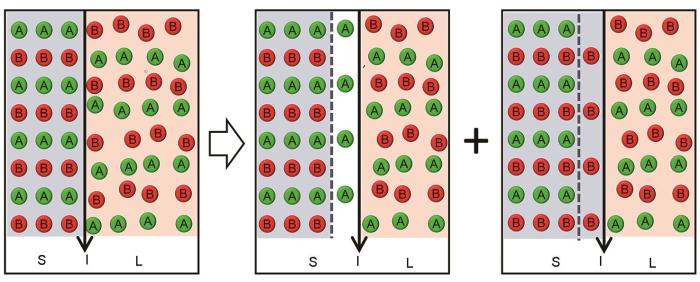
Schematics for solute trans-interface diffusion in one step (a) and two steps (b) (For the former, there is only one flux for solute trans-interface diffusion, i.e., = = , whereas for the latter, there are two fluxes for solute trans-interface diffusion, i.e., = = and = = .JD—the flux of solute trans-interface diffusion, —the flux of trans-interface diffusion from solid phase to interface, —the flux of trans-interface diffusion from interface to liquid phase, —the solute component at the interface that finally crosses the interface and enters the solid phase, V—the velocity of the migrating interface, Vm—mole volume)
Fig.6
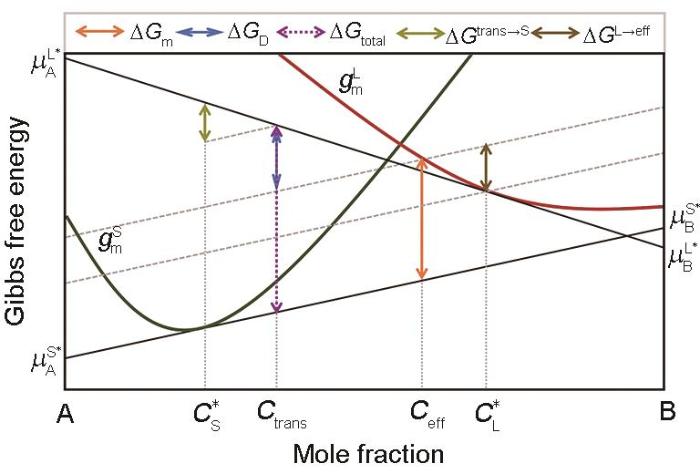
Mole Gibbs free energy diagram for the solid/liquid interface kinetic processes with a partial solute-drag effect under a non-steady-state condition (The total Gibbs free energy dissipated by the interface after solidification of 1 mol liquid is = . By translating the tangent of Gibbs free energy curve of solid at to the Gibbs free energy curve of liquid at Ceff, Gtotal is divided into two parts. The upper one for trans-interface diffusion is = and latter part for interface migration is Gm = . The difference in GD between the steady-state condition and the non-steady-state condition is = , which is the Gibbs free energy dissipated to adjust the actual composition transferred across the interface from to Ctrans for trans-interface diffusion. The difference in Gm between the two cases with a full solute-drag effect and with a partial solute-drag effect is = , which is the Gibbs free energy dissipated to adjust the liquid composition ahead of the solid/liquid interface from to Ceff for interface migration. It should be pointed out that Hareland et al. [30] confused the concepts of Ctrans under non-steady-state and Ceff. Furthermore, it is also queried that the definition of Ceff can be stilled be used for a non-steady state condition. For example, there will be a problem when < Ceff < Ctrans (Ceff—effective concentration)
The distribution characteristics and magnitude of energy density on the cross section of a laser beam are determined by its spatial profile, which directly impacts heat transport during laser material processing. Hence, it is essential to understand the influence of spatial profiles on heat transport during laser directed energy deposition with synchronous material delivery. Herein, a three-dimensional heat transport model that takes into account important physical events such as laser-powder-pool coupling, thermal-fluid coupling, solid-liquid phase change, and multiple heat transfer was established. The model was validated using single-track single-layer deposition experiments. The effects of four spatial laser beam profiles, including Gaussian (GP), super-Gaussian (SGP1 and SGP2), and pure flat-topped (FTP) profiles, on the heat transport and fluid flow within the molten pool were investigated. Simulated results show that peak temperatures of the molten pool decrease sequentially under GP, SGP1, SGP2 and FTP, and the temperature gradients on the solidification interface increase gradually from the top to the bottom of the molten pool. Temperature gradients on the solidification interface positively correlate with the angle between the normal direction of the solidification interface and the laser scanning direction, and negatively correlate with the distances from the beam center on the molten pool surface. Under all four spatial laser beam profiles, temperature gradients at the same positions on the solidification interface near the rear of the molten pool increase, while those at the bottom of the molten pool decrease. The molten pool exhibits an outward annular flow pattern under all four spatial laser beam profiles with fluid flows mainly driven by Marangoni shear stress. Heat transfer within the molten pool is dominated by Marangoni convection and heat conduction. Average fluid velocities within the molten pool decrease successively according to the following order: Gaussian, super-Gaussian, and pure flat-topped profiles.
任 松, 吴家柱, 张 屹等.
激光束空域形态对激光定向能量沉积316L不锈钢热输运影响的数值模拟
[J]. 金属学报, 2024, 60: 1678
WangY Q, FuK, ZhaoY Z, et al.
Non-equilibrium solidification behavior and microstructure evolution of undercooled Fe7(CoNi-Mn)80B13 eutectic high-entropy alloy
[J]. Acta Metall. Sin., 2025, 61: 143
王叶青, 付 珂, 赵永柱等.
Fe7(CoNiMn)80B13共晶高熵合金的深过冷非平衡凝固行为及微观组织演变
[J]. 金属学报, 2025, 61: 143
HuB, ZhangH Q, ZhangJ, et al.
Progress in interfacial thermodynamics and grain boundary complexion diagram
Grain boundaries (GBs), a crucial component of microstructures, have a significant influence on the properties of materials. The GB complexion (GBC) transitions are essential information to accurately explain numerous material phenomena. However, owing to the complexity of GB structures and the difficulty in observation of GBC transitions, there is still no direct evidence and mechanism explanation for these material phenomena. With the advancement of characterization equipment, especially spherical aberration-correction transmission electron microscopy, coupled with powerful computer simulation, the establishment of interfacial thermodynamic models to construct different types of GBC diagrams, which provide a broad prospect for the study of GB structures and GBC transitions, is essential. In this paper, the progress of interface thermodynamics and GBC diagrams from the aspects of the classification and characterization of GBs and GBC transitions, interface thermodynamic models, and the construction of GBC diagrams were reviewed. The paper also looks forward to the future development of interface thermodynamics and GBC diagrams.
Cu is commonly used in the field of electricity and electronics because of its high ductility, and electrical and thermal conductivity. The thermophysical properties and the atomic structure of liquid Cu, especially for undercooled state, are of practical significance in both application and fundamental researches. The major approaches to obtain thermophysical properties of undercooled metals are containerless techniques based on electrostatic levitation, electromagnetic levitation and ultrasonic levitation et al. However, the strong volatility of liquid Cu results in great difficulties to measure the thermophysical properties. Accordingly, computational prediction is becoming an expected method to obtain the thermophysical data of liquid Cu. The molecular dynamics (MD) simulation, in combination with a resonable potential model, has been extensively employed in studying the physical properties of several metals as a powerful approach. In this work, the atomic distribution and thermophysical properties including melting temperature, density, specific heat and self-diffusion coefficient of liquid Cu were studied by molecular dynamics simulation. Mishin's and Zhou's embedded-atom method potentials, and the modified embedded-atom method potential proposed by Baskes were used over the temperature range of 800~2400 K, reaching the maximum undercooling of 556 K. The simulated results are in good agreement with the reported experimental results. The crystal-liquid-crystal sandwich structure has been used to calculate the melting point. The melting point calculated by Baskes' potential model is 1341 K, just a difference of 1.11% from the experimental value. The density at the melting point calculated by Mishin's potential is 7.86 g/cm3, with a difference less than 2% compared with the reported data. It is found that the enthalpy of liquid Cu increases linearly with the increase of temperature. The specific heat is obtained to be 31.89 J/(molK) by Mishin's potential, which is constant in the corresponding temperature range. The self-diffusion coefficient is exponentially dependent on the temperature. The maximum error between the reported value and the present value of the self-diffusion coefficient calculated by Mishin's potential is only 4.93%. The pair distribution function was applied to investigate the atomic structure of liquid Cu, which suggests that the simulated system is still ordered in short range and disordered in long range for both normal liquid and undercooled state. It is found that the atomic ordered degree is weakened with the increase of temperature, and it is kept within 3~4 atom neighbor distance.
Rapid solidification is a relevant physical phenomenon in material sciences, whose theoretical analysis requires going beyond the limits of local equilibrium statistical physics and thermodynamics and, in particular, taking account of ergodicity breaking and of generalized formulation of thermodynamics. The ergodicity breaking is related to the time symmetry breaking and to the presence of some kinds of fluxes and gradient flows making that an average of microscopic variables along time is different than an average over some chosen statistical ensemble. In fast processes, this is due, for instance, to the fact that the system has no time enough to explore the whole region of possible microscopic states in the phase space. Similarly to this, systems submitted to strong fluxes may have no time for reaching the whole phase space in local bulks during observable macroscopic time. Rapid solidification, ergodicity breaking and extended thermodynamics actually make a conceptually novel combination in the present overview: ergodicity breaking is expressed in general terms and then extended thermodynamics is formulated as a particular phenomenological expression and applied to describe the dynamics of the phenomenon. Using the formalism of micro- and meso-scopic dynamics we introduce a general view on non-ergodic fast transitions and provide a simplest description of a continuum theory based on the system of hyperbolic equations applicable to rapid solidification. Analysis of non-equilibrium effects, including interface kinetics, solute trapping and solute drag, is presented with their effect on the rapidly moving solid-liquid interface. Special attention is paid to the theory predictions compared with the kinetics obtained in experiments on samples processed by electromagnetic levitation facility and in molecular dynamics simulation. (C) 2019 Elsevier B.V.
SobolevS L.
Local-nonequilibrium model for rapid solidification of undercooled melts
Especially in the past decades, Ti-6Al-4V alloy has received much attention, not only due to its high melting temperature, good corrosion resistance, low density and high hardness, but also because of the diverse and complicated microstructures formed under different conditions. This makes Ti-6Al-4V a potential candidate in both aerospace industries and fundamental research. It is well known that the solidified microstructures of alloy have a great influence on their mechanical properties. Therefore, it is crucial to investigate the mechanical properties of Ti-6Al-4V solidified under different conditions, in particular in the undercooling conditions. However, it is noted that most research on the solidification of Ti-6Al-4V alloy was carried out under equilibrium condition. With respect to Ti-6Al-4V alloy solidified under substantial undercooling conditions, few studies could be found. Thus, it is interesting to study two points: (1) the feature of the microstructure of Ti-6Al-4V alloy solidified under highly undercooled conditions and large cooling rate, (2) the influence of undercooling and cooling rate on the mechanical property of Ti-6Al-4V alloy. To address these two problems, Ti-6Al-4V alloy was rapidly solidified in a drop tube. The main results are summarized as follows. The microstructure of the Ti-6Al-4V alloy solidified under free fall condition displays "lamellar α+β→α dendrites→basket-weave α'+β→ needle-like α'→ needle-like α'+ anomalous β " transformation with decreasing the droplets diameter. And the needle-like α' phase in the original boundaries of equiaxed β grains is transformed into a continuous distribution and anomalous structure of β phase when the droplet size is less than about 400 μm. The microhardness of this alloy ranges from 506 kg/mm2 to 785 kg/mm2 when the droplet diameter decreases from 1420 μm to 88 μm, which is much higher than that of the master alloy. For "lamellar structure of α+β phases", "needle-like α' phase" and "needle-like α' phase+ anomalous β phase", the microhardness increases with the decrease of droplet diameter. But for 'basket-weave' microstructure, the microhardness diminishes with the decrease of droplet diameter.
Application of the thermodynamic extremal principle to diffusion-controlled phase-transformations in multi-component substitutional alloys: Modeling and applications
Selective laser melting (SLM) is a laser-based additive manufacturing technique that can fabricate parts with complex geometries and sufficient mechanical properties. However, the optimal SLM process windows of metallic materials are difficult to predict, especially when exploring new metallic materials. In this paper, a universal and simplified model has been proposed to predict the energy density suitable for SLM of a variety of metallic materials including Ti and Ti alloys, Al alloy, Ni-based superalloy and steel, on the basis of the relationship between energy absorption and consumption during SLM. Several important but easily overlooked factors, including the surface structure of metallic powder, porosity of powder bed, vaporization and heat loss, were considered to improve the accuracy of the model. Results show that, to achieve near-full density parts, the energy absorption (Q(a)) by the local powder bed should be approximately 3-8 times greater than the energy consumption (Q(c))and this finding applies to all materials investigated. The value of Q(a)/Q(c) highly depends on material properties, particularly laser absorptivity, latent heat of melting and specific heat capacity. Experiments on high-entropy alloy (CrMnFeCoNi) and Hastelloy X alloy, new metallic materials for SLM, have been further conducted to verify the model. Results confirm that the model can predict suitable laser energy densities needed for processing the various metallic materials without tedious trial and error experiments. Indications and uncertainty of the model have also been analyzed.
GuJ, JuJ, WangR, et al.
Effects of laser scanning rate and Ti content on wear of novel Fe-Cr-B-Al-Ti coating prepared via laser cladding
[J]. J. Therm. Spray Technol., 2022, 31: 2609
JuJ, YuH Y, ZhaoY L, et al.
Understanding the oxidation behaviors of a Ni-Co-based superalloy at elevated temperatures through multiscale characterization
[J]. Corros. Sci., 2024, 227: 111800
YangT, ZhaoY L, FanL, et al.
Control of nanoscale precipitation and elimination of intermediate-temperature embrittlement in multicomponent high-entropy alloys
Thermally stable high-entropy alloys (HEAs) consisting of a high density of coherent precipitates show a great potential for high-temperature applications. In this work, we systematically investigated the phase stability and coarsening kinetics of L1(2)-type coherent precipitates in a Ni-30Co-13Fe-15Cr-6Al-6Ti-0.1B (at.%) HEA isothermally aged at 800, 900 and 1000 degrees C. Aged microstructures in the grain interiors under this temperature range were essentially dominated by the uniform precipitation of multicomponent L1(2) (Ni, Co, Fe, Cr)(3)(Ti, Al)-type precipitates. The coarsening kinetics of these intragranular L1(2) precipitates were quantitatively determined, which were adequately characterized by the classical Lifshitz-Slyozov-Wagner model. The activation energy for coarsening was determined to be 378 kJ/mol, which is relatively higher than that of conventional Ni or Co-based superalloys, suggesting a slow elemental diffusion in the HEA matrix. More importantly, the heterogeneous precipitation and the associated metastable phase transformation mechanism along grain boundaries (GBs) were carefully analyzed. Localized chemical heterogeneity was identified within the discontinuous L1(2) phase at the GBs, which thermodynamically destabilizes the L1(2) structure and encourages the formation of brittle Heusler phase. Finally, we establish a unique duplex-aging strategy that can be efficiently utilized for GB stabilization, by which these detrimental intergranular heterostructures can be greatly eliminated, leading to an exceptional resistance to intermediate-temperature embrittlement, along with enhanced tensile strengths. These findings will not only shed light on the precipitation mechanisms in compositionally complex HEAs but also generate new opportunities to the interfacial design of HEAs for advanced high-temperature applications with superior properties. (C) 2020 Acta Materialia Inc. Published by Elsevier Ltd.
ZhangJ B, WangH F, KuangW W, et al.
Rapid solidification of non-stoichiometric intermetallic compounds: Modeling and experimental verification
The grain refinement mechanism for rapid solidification of undercooled melts is still an open problem even after 60 years of on-going studies. In this work, rapid solidification of undercooled Ni and equi-atomic FeCoNiPd melts was studied and spontaneous grain refinement was found at both low and high undercooling. After a detailed electron backscattered diffraction analysis, subgrain-induced grain orientation scattering and splitting were found to occur along with the transition from coarse dendrites to fine equiaxed grains at low and high undercooling, respectively, indicating that subgrains play an important role during the formation of fine equiaxed grains. On this basis, a compromise mechanism of subgrain-assisted spontaneous grain refinement was proposed. Because the dendrite re-melting induced thermo-mechanical process and fluid flow induced dendrite deformation occur simultaneously during the post-recalescence stage, stress accumulation would be maximum at both low and high undercooling, thus inducing dynamic recrystallization, during which the formation and rotation of subgrains make the grain orientations scattering and even splitting. Furthermore, the grain/subgrain size of undercooled FeCoNiPd ascribing to its unique co-segregation behavior keeps almost invariable from low to high undercooling, indicating that the co-segregation strategy would be effective to inhibit grain growth after rapid solidification and would be useful in practice.
CuiD X, ZhangJ B, LiX, et al.
Atomistic insights into sluggish crystal growth in an undercooled CoNiCrFe multi-principal element alloy
Application of the thermodynamic extremal principle to diffusion-controlled phase-transformations in multi-component substitutional alloys: Modeling and applications
... [30] confused the concepts of Ctrans under non-steady-state and Ceff. Furthermore, it is also queried that the definition of Ceff can be stilled be used for a non-steady state condition. For example, there will be a problem when < Ceff < Ctrans (Ceff—effective concentration)Fig.6<strong>4</strong> 结论与展望
本文以二元合金凝固为例,分析总结了非平衡界面动力学理论的发展现状和发展趋势.主要结论如下. ...
Phase-field modeling of an abrupt disappearance of solute drag in rapid solidification






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}