Ferroelectric materials, which are characterized by tunable spontaneous polarization, show remarkable application potential for nonvolatile information storage; however they present various challenges. The performance of these materials is strongly influenced by their dynamic polarization behavior under multiple external fields. Due to the limited precision of experimental observations, precise atomic-level material simulations are crucial. Although molecular dynamics (MD) offers an ideal method for investigating material dynamics over a wide spatiotemporal range, its application to new materials is often limited by challenges such as low accuracy, complex development, and limited portability of conventional classical force fields. Advances in machine learning have provided new possibilities for developing force fields. Among different machine learning potentials, deep potential (DP) based on deep neural networks stands out. DP offers accuracy comparable to that of density functional theory while providing computational efficiency similar to that of conventional classical force fields. This review primarily focused on the development and application of DP in ferroelectric materials, specifically examining the phase transition mechanisms and polarization reversal processes at the atomic scale. Considerable efforts have been made to develop and evaluate DP for crucial ferroelectric materials such as hafnium dioxide (HfO2) and classic perovskite ferroelectric materials. Furthermore, this review explores the high oxygen-ion migration kinetics in HfO2 using DP and investigates the flexoelectricity induced by polar domain boundaries and the bulk photovoltaic effects in strontium titanate. By highlighting the use of DP molecular dynamics approaches in ferroelectric materials, this review emphasizes the role of machine learning approaches in optimizing and accelerating material simulations to facilitate further breakthroughs and discoveries.
Keywords:ferroelectric material;
molecular dynamics;
machine learning;
deep potential
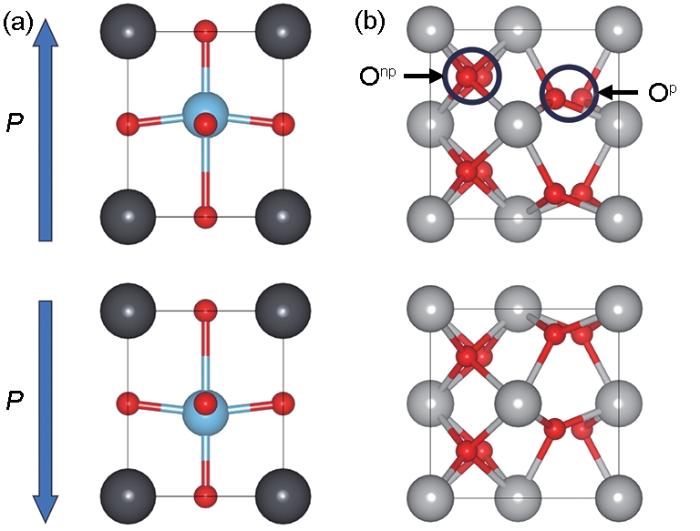
Fig.1
Schematic structures of perovskite ferroelectric material represented by PbTiO3 (Polarization (denote as P) directions are marked with blue arrows. Pb atoms in dark gray, Ti atoms in blue, and O atoms in red) (a) and ferroelectric phase HfO2 (Hf atoms in light gray and O atoms in red. Onp and Op are non-polar and polar O atoms, respectively) (b)
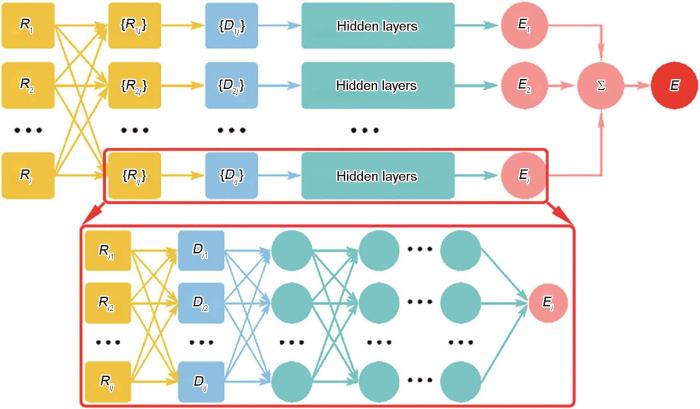
Fig.2
Schematic diagram of the deep potential (DP) energy model, amplified by a deep neural network [98] ( Ri is the global coordinates of the atom i, and Rij = Ri - Rj describes the neighboring atoms. Dij is the local coordinate information and serves as neural network input. Ei is the “atomic energy” of atom i, while E is the total energy of the whole system)
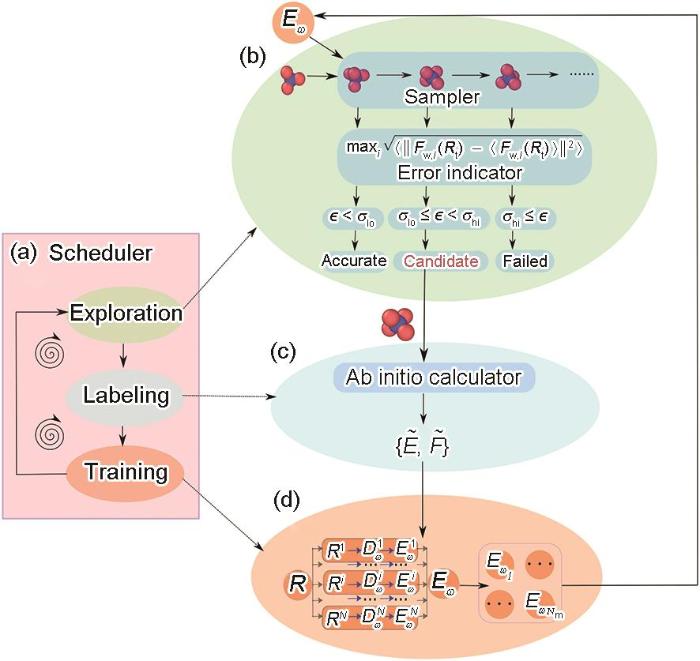
Fig.3
Deep potential generator (DP-GEN) framework diagram[86]
(a) the exploration, labeling, and training steps in the iterative procedure; the deep potential molecular dynamics (DPMD)-based explora-tion strategy is used here as an example. The molecular dynamics simulation is driven by an ensemble of DP models given an initial structure, and a series of conforma-tions are sampled
(b) for each configuration, the maximum deviation from the atomic force, defined as the error metric ϵ, is predicted by the DP model ensemble. σhi and σlo are the upper and lower bounds of the trust levels, respectively. Fw,i (Rt) denotes the force on the atom with index i predicted by the model Eω, and Nm denotes the number of atoms
(c) labeling is performed, and first-principles cal-culations are employed to obtain the energies and forces of the candidate structures, and are energies and forces computed through first-principles calculations, respectively
(d) based on the accumulated training set, a new DP model ensemble is generated and passed to the next iteration
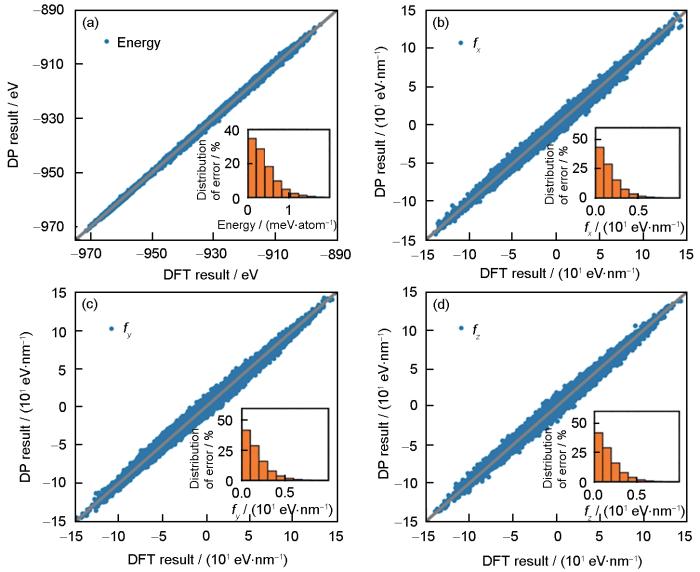
Fig.4
DP model predictions of energies (a) and atomic forces along different directions (fx, fy, and fz ) (b-d) for all structures in the final training dataset vs density functional theory (DFT) results (Insets represent absolute error distributions)[103]
图5a[103]展示了使用DP模型预测和DFT计算的HfO2不同晶相的状态方程(equations of state,EOS)。DP模型很好地预测了DFT计算的状态方程以及不同晶相稳定性的顺序:E(P21/c) < E(Pbca) < E(Pca21) < E(P42/nmc) < E(Fmm)。图5b[103]中展示的P21/c、Pbca和Pca21相的声子谱进一步证明了DP模型预测力学性能的能力,DP模型预测的声子谱结果与DFT计算值之间存在非常好的一致性。此外,向训练数据集中添加用于计算声子谱的扰动结构可以进一步改善DP模型对声子谱的预测能力。在HfO2薄膜中,铁电相(空间群Pca21)的形成被认为是动力学过程诱导的,但要研究有限温度下的相变,力场需要准确地预测不同晶相之间的能垒。由于相变过程的中间结构相对平衡结构发生了强烈的畸变,对于力场的精度和扩展性有较高的要求。USPEX代码[104]中的变胞微动弹性带(variable cell nudged elastic band,VCNEB)方法可以确定连接HfO2不同相之间的最小能量路径(minimum energy paths,MEPs)。图5c[103]对比了DP预测和DFT计算MEPs中结构的能量,显示了2者之间的出色一致性,平均绝对误差为2.2 meV/atom。
Fig.5
Equation of state for different crystalline phases of HfO2 (V is the volume of the cell. Solid lines and crosses mark the results of DFT calculations and DP model predictions, respectively)[103] (a), phonon spectra of HfO2P21/c, Pbca, and Pca21phases[103] (b), energy barrier calculations between multiple crystalline phases of HfO2 (Solid lines are DFT calculations, and hollow circles are DP predictions)[103] (c), probability distributions of the local displacement of oxygen atoms along the [010] direction (d[010]) at different temperatures for a pressure of 0 GPa (Inset shows the distribution of the displacements of oxygen atoms along the [100], [010], and [001] directions at 400 K) (d), and temperature increase-driven changes in the lattice constant and in the average value of d[010], indicating the occurrence of a phase transition (e)
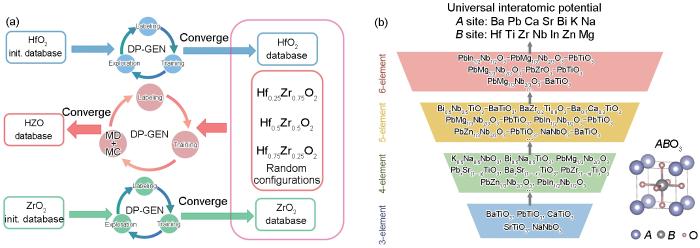
对于研究最为深入的ABO3型钙钛矿氧化物而言,其A位和B位的多样性为微调各种相互作用的能量尺度提供了平台,进而产生了丰富的新现象,如铁电性、铁磁性、多铁性和超导性等。Wu等[105]采用“模块化开发深度势”(modular development of deep potential,ModDP)的策略,系统地开发、扩展和改善了多组分固溶体的力场。ModDP的核心思想是将DP模型相关的训练数据集视为基本模块,这些模块可被重复利用,构建固溶体的初始训练数据集。然后,通过分子动力学模拟和Monte Carlo交换过程对其庞大的构型和化学空间进行采样,采用并同步学习程序DP-GEN自动、高效地迭代更新固溶体的训练数据集。如图6a[105]所示,ModDP策略可在没有人为干预的情况下直接开发复杂固溶体的DP模型,为建立模块化的力场数据集奠定了基础。进一步将ModDP策略应用于常见的ABO3类型的钙钛矿固溶体材料,结合具有注意力机制的神经网络模型DPA-1[106],获得了一个通用的力场,能够准确地描述包含14种不同金属元素的26种不同类型的钙钛矿型氧化物[107]。
Fig.6
Schematic diagram of modular development of deep potential (ModDP) strategy (As an example, DP-GEN is used to obtain DP models for HfO2 and ZrO2, respectively, using Hf x Zr1 - x O2 (HZO). The initial training dataset consists of the converged datasets of HfO2 and ZrO2 with the random structures of Hf x Zr1 - x O2 (x = 0.25, 0.50, and 0.75), and the converged DP model of Hf x Zr1 - x O2 is finally obtained by DP-GEN. MD—molecular dynamics, MC—Monte Carlo) (a)[105] and schematic diagram of the UniPero process (The workflow for building a generic force field for chalcogenide oxides follows the strategy of ModDP) (b)[107]
基于ModDP策略,Wu等[107]设计了一个逐步增加复杂性的程序,旨在增强DPA-1模型对钙钛矿型氧化物的描述能力。力场开发程序的工作流程如图6b[107]所示,该方法旨在获得通用的钙钛矿型氧化物原子间力场。初始数据集包含约1000个结构,涵盖26种不同类型的钙钛矿氧化物,涉及约200个组分和14种金属元素。首先,使用标准的DP-GEN策略来收敛3种元素的钙钛矿氧化物的DPA-1模型。然后,将收敛的训练数据集作为DP-GEN的起点,以获得适用于4种元素钙钛矿体系的DPA-1模型,例如PbZr1 - x Ti x O3和Pb(Mg1/3Nb2/3)O3。最终得到一个能够描述包括Pb(In1/2Nb1/2)O3-Pb(Mg1/3Nb2/3)O3-PbTiO3 (即PIN-PMN-PT)等三元固溶体在内的6种元素的钙钛矿型氧化物的DPA-1模型,该模型即为钙钛矿型铁电材料通用势场,命名为UniPero,能够描述多种钙钛矿型氧化物。
在确认模型能量和力准确收敛的前提下,DPA-1模型适用性可通过模拟钙钛矿型铁电材料的温度驱动相变来进行测试。如图7[107]所示,DPA-1模型在没有任何调整的情况下,成功地复现了铅基铁电体(如PbTiO3、Pb0.5Sr0.5TiO3和PbZr0.5Ti0.5O3)、无铅铁电体(如BaTiO3、KNbO3和K0.5Na0.5NbO3)、量子顺电体SrTiO3以及三元固溶体(0.29PIN-0.45PMN-0.26PT和0.36PIN-0.36PMN-0.28PT)温度驱动的相变过程,模拟结果与实验结果具有很好的一致性。基于NPT系综的分子动力学模拟得到温度与晶格常数的关系显示:在PbTiO3中,模拟预测了从四方晶系(T,空间群P4mm)向立方晶系(C,空间群Pmm)的相转变,相变温度(Tc)约为550 K (图7a[107]),而在A位掺杂的Pb0.5Sr0.5TiO3固溶体中,Tc降至约280 K (图7d[107])。相比之下,使用DPA-1模型模拟的B位掺杂的PbZr0.5Ti0.5O3固溶体,在低温下为菱形结构(R,空间群R3m),随着温度的升高发生了菱形-四方-立方(R-T-C)的相变(图7g[107]),与实验结果一致。量子顺电体SrTiO3随着温度的升高表现出四方-立方相变(I4/mcm→Pmm),其特征是TiO6八面体倾斜角的减小。这个相变对应着一个约为1 meV/atom的微小能垒,DPA-1模型成功地复现了该相变过程(图7b[107]),理论上的Tc值为150 K,与实验值105 K相当。此外,DPA-1模型还正确预测了BaTiO3和K0.5Na0.5NbO3中的相变过程,即菱形-正交-四方-立方(R-O-T-C) (图7c和h[107])。对于像PIN-PMN-PT这样复杂的包含6种元素的固溶体,DPA-1不仅能够复现实验中观察到的T-C相变,还能够正确预测由组分变化导致的Tc的变化趋势。如图7f和i[107]所示,0.29PIN-0.45PMN-0.26PT到0.36PIN-0.36PMN-0.28PT相变温度约升高50 K,类似于实验中PIN的组分含量从29%增加到36%,而Tc相应升高约60 K。值得说明的是,铁电相变的理论Tc值通常比实验值低100~200 K。这种差异是通过拟合PBEsol密度泛函计算结果所开发的力场的常见特征,考虑到DPA-1模型预测结果与DFT计算结果表现出的高度的一致性,Tc的低估更可能是由于PBEsol在预测有限温度方面具有局限性,而不是DPA-1模型精度的问题。
Fig.7
Correlation of temperature with lattice constant based on generalized interatomic potential simulations (T—tetragonal, C—cubic, R—rhombohedral, O—orthorhombic)
Fig.8
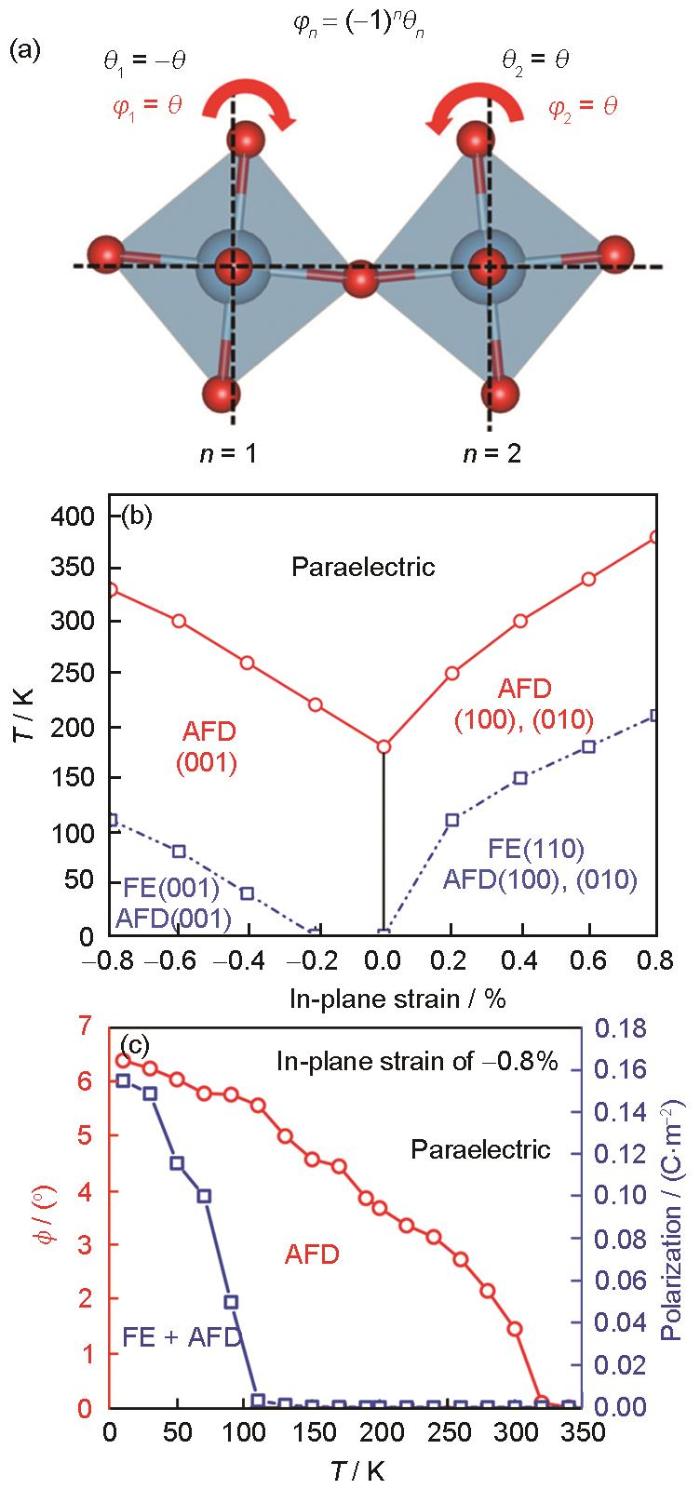
Schematic representation of the inverse ferroelectric distortion octahedral turning angle (φ) (φn is defined as the antiferrodistortive (AFD) order parameter, where the index n is the sequence number of unit cell, and θ is the rotation angle of TiO6 octahedra in each unit cell) (a), phase diagram of the SrTiO3 (STO) block under biaxial in-plane strain simulated by deep potential molecular dynamics (The ferroelectric (FE) and AFD transition temperatures at different strains are indicated by blue square dots and red dots, respectively. T represents temperature) (b), and curves of polarization and octahedral turning angle (ϕ) versus temperature for STO supercells (5000 atoms) under biaxial in-plane strain (-0.8%) (c)[116]
Fig.9
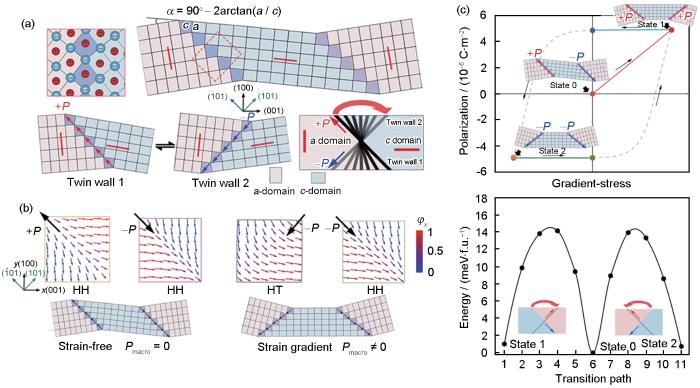
Schematic of the domain lattice point model in SrTiO3 (STO) films (Where the domains generate spontaneous concave and convex undulations in the (001) plane, with the convex angle corresponding to the domains with upward polarization and the concave angle corresponding to the domains with downward polarization, and the domains can be oriented to rotate in order to flip the polarization. a and c are lattice parameters, and α is the angel between a-c domain) (a), schematic diagram of a 90° rotation of polarized domain boundaries by bending deformation (Pmacro is the macroscopic polarization, head-to-tail and head-to-head domain configurations are denoted as HT and HH, respectively) (b), and hysteresis-like features of the STO film under strain, and after the stress is removed, the STO can still maintain the bent state with residual polarization (c)[117]
Fig.10
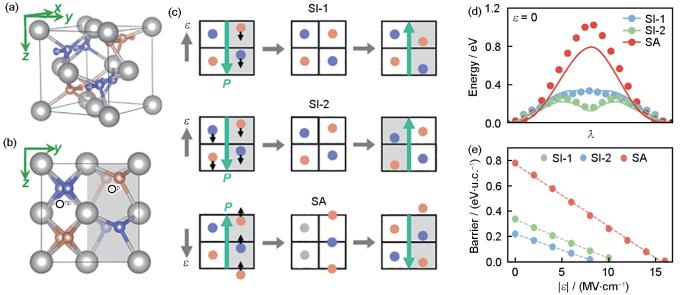
Polarization reversal paths in the ferroelectric phase HfO2 [128]
(a) vibrational modes of crystals in a single cell of Pca21, with outwardly and inwardly shifted oxygen atoms indicated by purple and brown-orange spheres, respectively, and polarization along the z-axis
(b) Pca21 has alternating arrangements of Onp and Op, with gray shaded regions marking the polar regions
(c) schematic representation of the shift in (SI) and shift across (SA) polarization reversal paths driven by an external electric field (ε). In the SA path, the Onp ion sign is reversed (indicated in grey during the transition). The SI and SA paths can start from the same structure, represented by P vectors in opposite directions (green arrows) to ensure compatibility with classical electrodynamics
(d) calculated results of the minimum energy paths for different polarization reversal paths, lines indicate the results of the DFT, and scatters indicate the results of the deep neural network-based force field prediction (λ represents the polariza-tion state (polar-nonpolar-polar))
(e) polarization reversal energy barrier versus electric field strength (|ε |)
Fig.11
Oxygen ion transport is coupled to the nucleation and growth mechanism of the ferroelectric polarization reversal[128]
(a) polar-antipolar phase cycling induced by successive SI and SA ferroelectric transitions. The highlighted marker Onp in the initial structure, δ = δ0 (defined as displacement of the Op ion relative to the top Hf plane), becomes Op after SI-2, and δ = -δ0, becomes Op after SA
(b) schematic representation of stochastic nucleation in a three-dimensional bulk. The nucleus is two-dimensional in the x-z plane and its thickness corresponds to half the cell length along the y-axis of the Pca21 phase of HfO2
(c) 2D-dimensional nucleus extracted from a DPMD simulation trace of a supercell containing 28800 atoms. The profile of the nucleus was determined based on the displacement δ-value of the Op ion
(d) δ profiles along the z and x directions labeled in Fig.11c
Perovskite light-emitting diodes with EQE exceeding 28% through a synergetic dual-additive strategy for defect passivation and nanostructure regulation
Photodetectors capture optical signals with a wide range of incident photon flux density and convert them to electrical signals instantaneously. They have many important applications including imaging, optical communication, remote control, chemical/biological sensing and so on. Currently, GaN, Si and InGaAs photodetectors are used in commercially available products. Here we demonstrate a novel solution-processed photodetector based on an organic-inorganic hybrid perovskite material. Operating at room temperature, the photodetectors exhibit a large detectivity (the ability to detect weak signals) approaching 10(14) Jones, a linear dynamic range over 100 decibels (dB) and a fast photoresponse with 3-dB bandwidth up to 3 MHz. The performance is significantly better than most of the organic, quantum dot and hybrid photodetectors reported so far; and is comparable, or even better than, the traditional inorganic semiconductor-based photodetectors. Our results indicate that with proper device interface design, perovskite materials are promising candidates for low-cost, high-performance photodetectors.
DeumelS, van BreemenA, GelinckG, et al.
High-sensitivity high-resolution X-ray imaging with soft-sintered metal halide perovskites
Resistive switching memory that uses halide perovskites (HP) has been considered as next-generation storage devices due to low operation voltage and high on/off ratio. However, the memory still faces challenges for stable operation with fast switching speed, which hinders the practical application. Thus, it should be considered from the stage of designing the HP for memory applications. Here, we design the perovskite memory using a high-throughput screening based on first-principles calculations. Total 696 compositions in four different crystal structures are investigated and essential parameters including stability, vacancy formation, and migration are considered as the descriptor. We select dimer-CsSbI as an optimal HP for memory; the device that uses dimer-CsSbI has ultra-fast switching speed (~20 ns) compared to the device that uses layer-CsSbI (>100 ns). The use of lead-free perovskite avoids environmental problems caused by lead in perovskite. These results demonstrate the feasibility to design the memory with ultra-fast switching speed.
ChoiJ, HanJ S, HongK, et al.
Organic-inorganic hybrid halide perovskites for memories, transistors, and artificial synapses
The unexpected ferroelectric properties of nanoscale hafnia-zirconia are considered to be promising for a wealth of applications including ferroelectric memory, field effect transistors, and energy-related applications. However, the reason why the unexpected ferroelectric Pca2 phase can be stabilized has not been clearly understood although numerous extensive theoretical and experimental results have been reported recently. The ferroelectric orthorhombic phase is not a stable phase under processing conditions from the viewpoint of bulk free energy. Although the possibility of stabilization of the ferroelectric phase due to the surface energy effect has been theoretically suggested, such a theoretical model has not been systematically compared with actual experimental results. In this study, the experimental observations on polymorphism in nanoscale HfO-ZrO solid solution thin films of a wide range of film compositions and thicknesses are comprehensively related to the theoretical predictions based on a thermodynamic surface energy model. The theoretical model can semi-quantitatively explain the experimental results on the phase-evolution, but there were non-negligible discrepancies between the two results. To understand these discrepancies, various factors such as the film stress, the role of a TiN capping layer, and the kinetics of crystallization are systematically studied. This work also reports on the evolution of electrical properties of the film, i.e. dielectric, ferroelectric, anti-ferroelectric, and morphotropic phase changes, as a function of the film composition and thickness. The in-depth analyses of the phase change are expected to provide an important guideline for subsequent studies.
XuL, NishimuraT, ShibayamaS, et al.
Ferroelectric phase stabilization of HfO2 by nitrogen doping
Memory devices with high speed and high density are highly desired to address the 'memory wall' issue. Here we demonstrated a highly scalable, three-dimensional stackable ferroelectric diode, with its rectifying polarity modulated by the polarization reversal of HfZrO films. By visualizing the hafnium/zirconium lattice order and oxygen lattice order with atomic-resolution spherical aberration-corrected STEM, we revealed the correlation between the spontaneous polarization of HfZrO film and the displacement of oxygen atom, thus unambiguously identified the non-centrosymmetric Pca2 orthorhombic phase in HfZrO film. We further implemented this ferroelectric diode in an 8 layers 3D array. Operation speed as high as 20 ns and robust endurance of more than 10 were demonstrated. The built-in nonlinearity of more than 100 guarantees its self-selective property that eliminates the need for external selectors to suppress the leakage current in large array. This work opens up new opportunities for future memory hierarchy evolution.
BouazizJ, Rojo RomeoP, BabouxN, et al.
Imprint issue during retention tests for HfO2-based FRAM: An industrial challenge?
[J]. Appl. Phys. Lett., 2021, 118: 082901
WuJ X, MoF, SarayaT, et al.
Monolithic integration of oxide semiconductor FET and ferroelectric capacitor enabled by Sn-doped InGaZnO for 3-D embedded RAM application
[J]. IEEE Trans. Electron Devices, 2021, 68: 6617
BeyerS, DünkelS, TrentzschM, et al.
FEFET: A versatile CMOS compatible device with game-changing potential
Deep learning is revolutionizing many areas of science and technology, especially image, text, and speech recognition. In this paper, we demonstrate how a deep neural network (NN) trained on quantum mechanical (QM) DFT calculations can learn an accurate and transferable potential for organic molecules. We introduce ANAKIN-ME (Accurate NeurAl networK engINe for Molecular Energies) or ANI for short. ANI is a new method designed with the intent of developing transferable neural network potentials that utilize a highly-modified version of the Behler and Parrinello symmetry functions to build single-atom atomic environment vectors (AEV) as a molecular representation. AEVs provide the ability to train neural networks to data that spans both configurational and conformational space, a feat not previously accomplished on this scale. We utilized ANI to build a potential called ANI-1, which was trained on a subset of the GDB databases with up to 8 heavy atoms in order to predict total energies for organic molecules containing four atom types: H, C, N, and O. To obtain an accelerated but physically relevant sampling of molecular potential surfaces, we also proposed a Normal Mode Sampling (NMS) method for generating molecular conformations. Through a series of case studies, we show that ANI-1 is chemically accurate compared to reference DFT calculations on much larger molecular systems (up to 54 atoms) than those included in the training data set.
ManzhosS, CarringtonT.
Using neural networks, optimized coordinates, and high-dimensional model representations to obtain a vinyl bromide potential surface
Whereas the interactions between water molecules are dominated by strongly directional hydrogen bonds (HBs), it was recently proposed that relatively weak, isotropic van der Waals (vdW) forces are essential for understanding the properties of liquid water and ice. This insight was derived from ab initio computer simulations, which provide an unbiased description of water at the atomic level and yield information on the underlying molecular forces. However, the high computational cost of such simulations prevents the systematic investigation of the influence of vdW forces on the thermodynamic anomalies of water. Here, we develop efficient ab initio-quality neural network potentials and use them to demonstrate that vdW interactions are crucial for the formation of water's density maximum and its negative volume of melting. Both phenomena can be explained by the flexibility of the HB network, which is the result of a delicate balance of weak vdW forces, causing, e.g., a pronounced expansion of the second solvation shell upon cooling that induces the density maximum.
KoH Y, ZhangL F, SantraB, et al.
Isotope effects in liquid water via deep potential molecular dynamics
Optimization and validation of a deep learning CuZr atomistic potential: Robust applications for crystalline and amorphous phases with near-DFT accuracy
Combustion is a complex chemical system which involves thousands of chemical reactions and generates hundreds of molecular species and radicals during the process. In this work, a neural network-based molecular dynamics (MD) simulation is carried out to simulate the benchmark combustion of methane. During MD simulation, detailed reaction processes leading to the creation of specific molecular species including various intermediate radicals and the products are intimately revealed and characterized. Overall, a total of 798 different chemical reactions were recorded and some new chemical reaction pathways were discovered. We believe that the present work heralds the dawn of a new era in which neural network-based reactive MD simulation can be practically applied to simulating important complex reaction systems at ab initio level, which provides atomic-level understanding of chemical reaction processes as well as discovery of new reaction pathways at an unprecedented level of detail beyond what laboratory experiments could accomplish.
AndradeM F C, KoH Y, ZhangL F, et al.
Free energy of proton transfer at the water-TiO2 interface from ab initio deep potential molecular dynamics
Perovskite light-emitting diodes with EQE exceeding 28% through a synergetic dual-additive strategy for defect passivation and nanostructure regulation
(a) the exploration, labeling, and training steps in the iterative procedure; the deep potential molecular dynamics (DPMD)-based explora-tion strategy is used here as an example. The molecular dynamics simulation is driven by an ensemble of DP models given an initial structure, and a series of conforma-tions are sampled ...
... [86]
(a) the exploration, labeling, and training steps in the iterative procedure; the deep potential molecular dynamics (DPMD)-based explora-tion strategy is used here as an example. The molecular dynamics simulation is driven by an ensemble of DP models given an initial structure, and a series of conforma-tions are sampled ...
Optimization and validation of a deep learning CuZr atomistic potential: Robust applications for crystalline and amorphous phases with near-DFT accuracy
Schematic diagram of the deep potential (DP) energy model, amplified by a deep neural network<sup> [<xref ref-type="bibr" rid="R98">98</xref>]</sup> ( <strong><i>R</i></strong><i><sub>i</sub></i> is the global coordinates of the atom <i>i</i>, and <strong><i>R</i></strong><i><sub>ij</sub></i> = <strong><i>R</i></strong><i><sub>i</sub></i> - <strong><i>R</i></strong><i><sub>j</sub></i> describes the neighboring atoms. <strong><i>D</i></strong><i><sub>ij</sub></i> is the local coordinate information and serves as neural network input. <i>E<sub>i</sub></i> is the “atomic energy” of atom <i>i,</i> while <i>E</i> is the total energy of the whole system)Fig.2
... [98] ( Ri is the global coordinates of the atom i, and Rij = Ri - Rj describes the neighboring atoms. Dij is the local coordinate information and serves as neural network input. Ei is the “atomic energy” of atom i, while E is the total energy of the whole system)Fig.2
... [103]DP model predictions of energies (a) and atomic forces along different directions (<i>f<sub>x</sub></i>, <i>f<sub>y</sub></i>, and <i>f<sub>z</sub></i> ) (b-d) for all structures in the final training dataset <i>vs</i> density functional theory (DFT) results (Insets represent absolute error distributions)<sup>[<xref ref-type="bibr" rid="R103">103</xref>]</sup>Fig.4
图5a[103]展示了使用DP模型预测和DFT计算的HfO2不同晶相的状态方程(equations of state,EOS).DP模型很好地预测了DFT计算的状态方程以及不同晶相稳定性的顺序:E(P21/c) < E(Pbca) < E(Pca21) < E(P42/nmc) < E(Fmm).图5b[103]中展示的P21/c、Pbca和Pca21相的声子谱进一步证明了DP模型预测力学性能的能力,DP模型预测的声子谱结果与DFT计算值之间存在非常好的一致性.此外,向训练数据集中添加用于计算声子谱的扰动结构可以进一步改善DP模型对声子谱的预测能力.在HfO2薄膜中,铁电相(空间群Pca21)的形成被认为是动力学过程诱导的,但要研究有限温度下的相变,力场需要准确地预测不同晶相之间的能垒.由于相变过程的中间结构相对平衡结构发生了强烈的畸变,对于力场的精度和扩展性有较高的要求.USPEX代码[104]中的变胞微动弹性带(variable cell nudged elastic band,VCNEB)方法可以确定连接HfO2不同相之间的最小能量路径(minimum energy paths,MEPs).图5c[103]对比了DP预测和DFT计算MEPs中结构的能量,显示了2者之间的出色一致性,平均绝对误差为2.2 meV/atom. ...
... [103]Fig.4
图5a[103]展示了使用DP模型预测和DFT计算的HfO2不同晶相的状态方程(equations of state,EOS).DP模型很好地预测了DFT计算的状态方程以及不同晶相稳定性的顺序:E(P21/c) < E(Pbca) < E(Pca21) < E(P42/nmc) < E(Fmm).图5b[103]中展示的P21/c、Pbca和Pca21相的声子谱进一步证明了DP模型预测力学性能的能力,DP模型预测的声子谱结果与DFT计算值之间存在非常好的一致性.此外,向训练数据集中添加用于计算声子谱的扰动结构可以进一步改善DP模型对声子谱的预测能力.在HfO2薄膜中,铁电相(空间群Pca21)的形成被认为是动力学过程诱导的,但要研究有限温度下的相变,力场需要准确地预测不同晶相之间的能垒.由于相变过程的中间结构相对平衡结构发生了强烈的畸变,对于力场的精度和扩展性有较高的要求.USPEX代码[104]中的变胞微动弹性带(variable cell nudged elastic band,VCNEB)方法可以确定连接HfO2不同相之间的最小能量路径(minimum energy paths,MEPs).图5c[103]对比了DP预测和DFT计算MEPs中结构的能量,显示了2者之间的出色一致性,平均绝对误差为2.2 meV/atom. ...
... 图5a[103]展示了使用DP模型预测和DFT计算的HfO2不同晶相的状态方程(equations of state,EOS).DP模型很好地预测了DFT计算的状态方程以及不同晶相稳定性的顺序:E(P21/c) < E(Pbca) < E(Pca21) < E(P42/nmc) < E(Fmm).图5b[103]中展示的P21/c、Pbca和Pca21相的声子谱进一步证明了DP模型预测力学性能的能力,DP模型预测的声子谱结果与DFT计算值之间存在非常好的一致性.此外,向训练数据集中添加用于计算声子谱的扰动结构可以进一步改善DP模型对声子谱的预测能力.在HfO2薄膜中,铁电相(空间群Pca21)的形成被认为是动力学过程诱导的,但要研究有限温度下的相变,力场需要准确地预测不同晶相之间的能垒.由于相变过程的中间结构相对平衡结构发生了强烈的畸变,对于力场的精度和扩展性有较高的要求.USPEX代码[104]中的变胞微动弹性带(variable cell nudged elastic band,VCNEB)方法可以确定连接HfO2不同相之间的最小能量路径(minimum energy paths,MEPs).图5c[103]对比了DP预测和DFT计算MEPs中结构的能量,显示了2者之间的出色一致性,平均绝对误差为2.2 meV/atom. ...
... [103]中展示的P21/c、Pbca和Pca21相的声子谱进一步证明了DP模型预测力学性能的能力,DP模型预测的声子谱结果与DFT计算值之间存在非常好的一致性.此外,向训练数据集中添加用于计算声子谱的扰动结构可以进一步改善DP模型对声子谱的预测能力.在HfO2薄膜中,铁电相(空间群Pca21)的形成被认为是动力学过程诱导的,但要研究有限温度下的相变,力场需要准确地预测不同晶相之间的能垒.由于相变过程的中间结构相对平衡结构发生了强烈的畸变,对于力场的精度和扩展性有较高的要求.USPEX代码[104]中的变胞微动弹性带(variable cell nudged elastic band,VCNEB)方法可以确定连接HfO2不同相之间的最小能量路径(minimum energy paths,MEPs).图5c[103]对比了DP预测和DFT计算MEPs中结构的能量,显示了2者之间的出色一致性,平均绝对误差为2.2 meV/atom. ...
... [103],HfO2P21/c、Pbca和Pca21相的声子谱[103],HfO2多种晶相间能垒计算[103],压力为0 GPa时不同温度下O原子沿[010]方向的局域位移(d[010])的概率分布,以及温度升高驱动的晶格常数以及d[010]平均值的变化[103]Equation of state for different crystalline phases of HfO<sub>2</sub> (<i>V</i> is the volume of the cell. Solid lines and crosses mark the results of DFT calculations and DP model predictions, respectively)<sup>[<xref ref-type="bibr" rid="R103">103</xref>]</sup> (a), phonon spectra of HfO<sub>2</sub><i>P</i>2<sub>1</sub>/<i>c</i>, <i>Pbca</i>, and <i>Pca</i>2<sub>1</sub>phases<sup>[<xref ref-type="bibr" rid="R103">103</xref>]</sup> (b), energy barrier calculations between multiple crystalline phases of HfO<sub>2</sub> (Solid lines are DFT calculations, and hollow circles are DP predictions)<sup>[<xref ref-type="bibr" rid="R103">103</xref>]</sup> (c), probability distributions of the local displacement of oxygen atoms along the [010] direction (<i>d</i><sub>[010]</sub>) at different temperatures for a pressure of 0 GPa (Inset shows the distribution of the displacements of oxygen atoms along the [100], [010], and [001] directions at 400 K) (d), and temperature increase-driven changes in the lattice constant and in the average value of <i>d</i><sub>[010]</sub>, indicating the occurrence of a phase transition (e)Fig.5
... [103],HfO2多种晶相间能垒计算[103],压力为0 GPa时不同温度下O原子沿[010]方向的局域位移(d[010])的概率分布,以及温度升高驱动的晶格常数以及d[010]平均值的变化[103]Equation of state for different crystalline phases of HfO<sub>2</sub> (<i>V</i> is the volume of the cell. Solid lines and crosses mark the results of DFT calculations and DP model predictions, respectively)<sup>[<xref ref-type="bibr" rid="R103">103</xref>]</sup> (a), phonon spectra of HfO<sub>2</sub><i>P</i>2<sub>1</sub>/<i>c</i>, <i>Pbca</i>, and <i>Pca</i>2<sub>1</sub>phases<sup>[<xref ref-type="bibr" rid="R103">103</xref>]</sup> (b), energy barrier calculations between multiple crystalline phases of HfO<sub>2</sub> (Solid lines are DFT calculations, and hollow circles are DP predictions)<sup>[<xref ref-type="bibr" rid="R103">103</xref>]</sup> (c), probability distributions of the local displacement of oxygen atoms along the [010] direction (<i>d</i><sub>[010]</sub>) at different temperatures for a pressure of 0 GPa (Inset shows the distribution of the displacements of oxygen atoms along the [100], [010], and [001] directions at 400 K) (d), and temperature increase-driven changes in the lattice constant and in the average value of <i>d</i><sub>[010]</sub>, indicating the occurrence of a phase transition (e)Fig.5
... [103],压力为0 GPa时不同温度下O原子沿[010]方向的局域位移(d[010])的概率分布,以及温度升高驱动的晶格常数以及d[010]平均值的变化[103]Equation of state for different crystalline phases of HfO<sub>2</sub> (<i>V</i> is the volume of the cell. Solid lines and crosses mark the results of DFT calculations and DP model predictions, respectively)<sup>[<xref ref-type="bibr" rid="R103">103</xref>]</sup> (a), phonon spectra of HfO<sub>2</sub><i>P</i>2<sub>1</sub>/<i>c</i>, <i>Pbca</i>, and <i>Pca</i>2<sub>1</sub>phases<sup>[<xref ref-type="bibr" rid="R103">103</xref>]</sup> (b), energy barrier calculations between multiple crystalline phases of HfO<sub>2</sub> (Solid lines are DFT calculations, and hollow circles are DP predictions)<sup>[<xref ref-type="bibr" rid="R103">103</xref>]</sup> (c), probability distributions of the local displacement of oxygen atoms along the [010] direction (<i>d</i><sub>[010]</sub>) at different temperatures for a pressure of 0 GPa (Inset shows the distribution of the displacements of oxygen atoms along the [100], [010], and [001] directions at 400 K) (d), and temperature increase-driven changes in the lattice constant and in the average value of <i>d</i><sub>[010]</sub>, indicating the occurrence of a phase transition (e)Fig.5
... [103]Equation of state for different crystalline phases of HfO<sub>2</sub> (<i>V</i> is the volume of the cell. Solid lines and crosses mark the results of DFT calculations and DP model predictions, respectively)<sup>[<xref ref-type="bibr" rid="R103">103</xref>]</sup> (a), phonon spectra of HfO<sub>2</sub><i>P</i>2<sub>1</sub>/<i>c</i>, <i>Pbca</i>, and <i>Pca</i>2<sub>1</sub>phases<sup>[<xref ref-type="bibr" rid="R103">103</xref>]</sup> (b), energy barrier calculations between multiple crystalline phases of HfO<sub>2</sub> (Solid lines are DFT calculations, and hollow circles are DP predictions)<sup>[<xref ref-type="bibr" rid="R103">103</xref>]</sup> (c), probability distributions of the local displacement of oxygen atoms along the [010] direction (<i>d</i><sub>[010]</sub>) at different temperatures for a pressure of 0 GPa (Inset shows the distribution of the displacements of oxygen atoms along the [100], [010], and [001] directions at 400 K) (d), and temperature increase-driven changes in the lattice constant and in the average value of <i>d</i><sub>[010]</sub>, indicating the occurrence of a phase transition (e)Fig.5
... [103] (a), phonon spectra of HfO2P21/c, Pbca, and Pca21phases[103] (b), energy barrier calculations between multiple crystalline phases of HfO2 (Solid lines are DFT calculations, and hollow circles are DP predictions)[103] (c), probability distributions of the local displacement of oxygen atoms along the [010] direction (d[010]) at different temperatures for a pressure of 0 GPa (Inset shows the distribution of the displacements of oxygen atoms along the [100], [010], and [001] directions at 400 K) (d), and temperature increase-driven changes in the lattice constant and in the average value of d[010], indicating the occurrence of a phase transition (e)Fig.5
... [103] (b), energy barrier calculations between multiple crystalline phases of HfO2 (Solid lines are DFT calculations, and hollow circles are DP predictions)[103] (c), probability distributions of the local displacement of oxygen atoms along the [010] direction (d[010]) at different temperatures for a pressure of 0 GPa (Inset shows the distribution of the displacements of oxygen atoms along the [100], [010], and [001] directions at 400 K) (d), and temperature increase-driven changes in the lattice constant and in the average value of d[010], indicating the occurrence of a phase transition (e)Fig.5
... [103] (c), probability distributions of the local displacement of oxygen atoms along the [010] direction (d[010]) at different temperatures for a pressure of 0 GPa (Inset shows the distribution of the displacements of oxygen atoms along the [100], [010], and [001] directions at 400 K) (d), and temperature increase-driven changes in the lattice constant and in the average value of d[010], indicating the occurrence of a phase transition (e)Fig.5
New developments in evolutionary structure prediction algorithm USPEX
1
2013
... 图5a[103]展示了使用DP模型预测和DFT计算的HfO2不同晶相的状态方程(equations of state,EOS).DP模型很好地预测了DFT计算的状态方程以及不同晶相稳定性的顺序:E(P21/c) < E(Pbca) < E(Pca21) < E(P42/nmc) < E(Fmm).图5b[103]中展示的P21/c、Pbca和Pca21相的声子谱进一步证明了DP模型预测力学性能的能力,DP模型预测的声子谱结果与DFT计算值之间存在非常好的一致性.此外,向训练数据集中添加用于计算声子谱的扰动结构可以进一步改善DP模型对声子谱的预测能力.在HfO2薄膜中,铁电相(空间群Pca21)的形成被认为是动力学过程诱导的,但要研究有限温度下的相变,力场需要准确地预测不同晶相之间的能垒.由于相变过程的中间结构相对平衡结构发生了强烈的畸变,对于力场的精度和扩展性有较高的要求.USPEX代码[104]中的变胞微动弹性带(variable cell nudged elastic band,VCNEB)方法可以确定连接HfO2不同相之间的最小能量路径(minimum energy paths,MEPs).图5c[103]对比了DP预测和DFT计算MEPs中结构的能量,显示了2者之间的出色一致性,平均绝对误差为2.2 meV/atom. ...
Modular development of deep potential for complex solid solutions
4
2023
... 对于研究最为深入的ABO3型钙钛矿氧化物而言,其A位和B位的多样性为微调各种相互作用的能量尺度提供了平台,进而产生了丰富的新现象,如铁电性、铁磁性、多铁性和超导性等.Wu等[105]采用“模块化开发深度势”(modular development of deep potential,ModDP)的策略,系统地开发、扩展和改善了多组分固溶体的力场.ModDP的核心思想是将DP模型相关的训练数据集视为基本模块,这些模块可被重复利用,构建固溶体的初始训练数据集.然后,通过分子动力学模拟和Monte Carlo交换过程对其庞大的构型和化学空间进行采样,采用并同步学习程序DP-GEN自动、高效地迭代更新固溶体的训练数据集.如图6a[105]所示,ModDP策略可在没有人为干预的情况下直接开发复杂固溶体的DP模型,为建立模块化的力场数据集奠定了基础.进一步将ModDP策略应用于常见的ABO3类型的钙钛矿固溶体材料,结合具有注意力机制的神经网络模型DPA-1[106],获得了一个通用的力场,能够准确地描述包含14种不同金属元素的26种不同类型的钙钛矿型氧化物[107]. ...
... [105]和UniPero 流程示意图[107]Schematic diagram of modular development of deep potential (ModDP) strategy (As an example, DP-GEN is used to obtain DP models for HfO<sub>2</sub> and ZrO<sub>2</sub>, respectively, using Hf <i><sub>x</sub></i> Zr<sub>1 - </sub><i><sub>x</sub></i> O<sub>2</sub> (HZO). The initial training dataset consists of the converged datasets of HfO<sub>2 </sub>and ZrO<sub>2</sub> with the random structures of Hf <i><sub>x</sub></i> Zr<sub>1 - </sub><i><sub>x</sub></i> O<sub>2</sub> (<i>x</i> = 0.25, 0.50, and 0.75), and the converged DP model of Hf <i><sub>x</sub></i> Zr<sub>1 - </sub><i><sub>x</sub></i> O<sub>2</sub> is finally obtained by DP-GEN. MD—molecular dynamics, MC—Monte Carlo) (a)<sup>[<xref ref-type="bibr" rid="R105">105</xref>]</sup> and schematic diagram of the UniPero process (The workflow for building a generic force field for chalcogenide oxides follows the strategy of ModDP) (b)<sup>[<xref ref-type="bibr" rid="R107">107</xref>]</sup>Fig.6
基于ModDP策略,Wu等[107]设计了一个逐步增加复杂性的程序,旨在增强DPA-1模型对钙钛矿型氧化物的描述能力.力场开发程序的工作流程如图6b[107]所示,该方法旨在获得通用的钙钛矿型氧化物原子间力场.初始数据集包含约1000个结构,涵盖26种不同类型的钙钛矿氧化物,涉及约200个组分和14种金属元素.首先,使用标准的DP-GEN策略来收敛3种元素的钙钛矿氧化物的DPA-1模型.然后,将收敛的训练数据集作为DP-GEN的起点,以获得适用于4种元素钙钛矿体系的DPA-1模型,例如PbZr1 - x Ti x O3和Pb(Mg1/3Nb2/3)O3.最终得到一个能够描述包括Pb(In1/2Nb1/2)O3-Pb(Mg1/3Nb2/3)O3-PbTiO3 (即PIN-PMN-PT)等三元固溶体在内的6种元素的钙钛矿型氧化物的DPA-1模型,该模型即为钙钛矿型铁电材料通用势场,命名为UniPero,能够描述多种钙钛矿型氧化物. ...
... [105] and schematic diagram of the UniPero process (The workflow for building a generic force field for chalcogenide oxides follows the strategy of ModDP) (b)[107]Fig.6
基于ModDP策略,Wu等[107]设计了一个逐步增加复杂性的程序,旨在增强DPA-1模型对钙钛矿型氧化物的描述能力.力场开发程序的工作流程如图6b[107]所示,该方法旨在获得通用的钙钛矿型氧化物原子间力场.初始数据集包含约1000个结构,涵盖26种不同类型的钙钛矿氧化物,涉及约200个组分和14种金属元素.首先,使用标准的DP-GEN策略来收敛3种元素的钙钛矿氧化物的DPA-1模型.然后,将收敛的训练数据集作为DP-GEN的起点,以获得适用于4种元素钙钛矿体系的DPA-1模型,例如PbZr1 - x Ti x O3和Pb(Mg1/3Nb2/3)O3.最终得到一个能够描述包括Pb(In1/2Nb1/2)O3-Pb(Mg1/3Nb2/3)O3-PbTiO3 (即PIN-PMN-PT)等三元固溶体在内的6种元素的钙钛矿型氧化物的DPA-1模型,该模型即为钙钛矿型铁电材料通用势场,命名为UniPero,能够描述多种钙钛矿型氧化物. ...
Pretraining of attention-based deep learning potential model for molecular simulation
1
2024
... 对于研究最为深入的ABO3型钙钛矿氧化物而言,其A位和B位的多样性为微调各种相互作用的能量尺度提供了平台,进而产生了丰富的新现象,如铁电性、铁磁性、多铁性和超导性等.Wu等[105]采用“模块化开发深度势”(modular development of deep potential,ModDP)的策略,系统地开发、扩展和改善了多组分固溶体的力场.ModDP的核心思想是将DP模型相关的训练数据集视为基本模块,这些模块可被重复利用,构建固溶体的初始训练数据集.然后,通过分子动力学模拟和Monte Carlo交换过程对其庞大的构型和化学空间进行采样,采用并同步学习程序DP-GEN自动、高效地迭代更新固溶体的训练数据集.如图6a[105]所示,ModDP策略可在没有人为干预的情况下直接开发复杂固溶体的DP模型,为建立模块化的力场数据集奠定了基础.进一步将ModDP策略应用于常见的ABO3类型的钙钛矿固溶体材料,结合具有注意力机制的神经网络模型DPA-1[106],获得了一个通用的力场,能够准确地描述包含14种不同金属元素的26种不同类型的钙钛矿型氧化物[107]. ...
Universal interatomic potential for perovskite oxides
13
2023
... 对于研究最为深入的ABO3型钙钛矿氧化物而言,其A位和B位的多样性为微调各种相互作用的能量尺度提供了平台,进而产生了丰富的新现象,如铁电性、铁磁性、多铁性和超导性等.Wu等[105]采用“模块化开发深度势”(modular development of deep potential,ModDP)的策略,系统地开发、扩展和改善了多组分固溶体的力场.ModDP的核心思想是将DP模型相关的训练数据集视为基本模块,这些模块可被重复利用,构建固溶体的初始训练数据集.然后,通过分子动力学模拟和Monte Carlo交换过程对其庞大的构型和化学空间进行采样,采用并同步学习程序DP-GEN自动、高效地迭代更新固溶体的训练数据集.如图6a[105]所示,ModDP策略可在没有人为干预的情况下直接开发复杂固溶体的DP模型,为建立模块化的力场数据集奠定了基础.进一步将ModDP策略应用于常见的ABO3类型的钙钛矿固溶体材料,结合具有注意力机制的神经网络模型DPA-1[106],获得了一个通用的力场,能够准确地描述包含14种不同金属元素的26种不同类型的钙钛矿型氧化物[107]. ...
... [107]Schematic diagram of modular development of deep potential (ModDP) strategy (As an example, DP-GEN is used to obtain DP models for HfO<sub>2</sub> and ZrO<sub>2</sub>, respectively, using Hf <i><sub>x</sub></i> Zr<sub>1 - </sub><i><sub>x</sub></i> O<sub>2</sub> (HZO). The initial training dataset consists of the converged datasets of HfO<sub>2 </sub>and ZrO<sub>2</sub> with the random structures of Hf <i><sub>x</sub></i> Zr<sub>1 - </sub><i><sub>x</sub></i> O<sub>2</sub> (<i>x</i> = 0.25, 0.50, and 0.75), and the converged DP model of Hf <i><sub>x</sub></i> Zr<sub>1 - </sub><i><sub>x</sub></i> O<sub>2</sub> is finally obtained by DP-GEN. MD—molecular dynamics, MC—Monte Carlo) (a)<sup>[<xref ref-type="bibr" rid="R105">105</xref>]</sup> and schematic diagram of the UniPero process (The workflow for building a generic force field for chalcogenide oxides follows the strategy of ModDP) (b)<sup>[<xref ref-type="bibr" rid="R107">107</xref>]</sup>Fig.6
基于ModDP策略,Wu等[107]设计了一个逐步增加复杂性的程序,旨在增强DPA-1模型对钙钛矿型氧化物的描述能力.力场开发程序的工作流程如图6b[107]所示,该方法旨在获得通用的钙钛矿型氧化物原子间力场.初始数据集包含约1000个结构,涵盖26种不同类型的钙钛矿氧化物,涉及约200个组分和14种金属元素.首先,使用标准的DP-GEN策略来收敛3种元素的钙钛矿氧化物的DPA-1模型.然后,将收敛的训练数据集作为DP-GEN的起点,以获得适用于4种元素钙钛矿体系的DPA-1模型,例如PbZr1 - x Ti x O3和Pb(Mg1/3Nb2/3)O3.最终得到一个能够描述包括Pb(In1/2Nb1/2)O3-Pb(Mg1/3Nb2/3)O3-PbTiO3 (即PIN-PMN-PT)等三元固溶体在内的6种元素的钙钛矿型氧化物的DPA-1模型,该模型即为钙钛矿型铁电材料通用势场,命名为UniPero,能够描述多种钙钛矿型氧化物. ...
... [107]Fig.6
基于ModDP策略,Wu等[107]设计了一个逐步增加复杂性的程序,旨在增强DPA-1模型对钙钛矿型氧化物的描述能力.力场开发程序的工作流程如图6b[107]所示,该方法旨在获得通用的钙钛矿型氧化物原子间力场.初始数据集包含约1000个结构,涵盖26种不同类型的钙钛矿氧化物,涉及约200个组分和14种金属元素.首先,使用标准的DP-GEN策略来收敛3种元素的钙钛矿氧化物的DPA-1模型.然后,将收敛的训练数据集作为DP-GEN的起点,以获得适用于4种元素钙钛矿体系的DPA-1模型,例如PbZr1 - x Ti x O3和Pb(Mg1/3Nb2/3)O3.最终得到一个能够描述包括Pb(In1/2Nb1/2)O3-Pb(Mg1/3Nb2/3)O3-PbTiO3 (即PIN-PMN-PT)等三元固溶体在内的6种元素的钙钛矿型氧化物的DPA-1模型,该模型即为钙钛矿型铁电材料通用势场,命名为UniPero,能够描述多种钙钛矿型氧化物. ...
... 基于ModDP策略,Wu等[107]设计了一个逐步增加复杂性的程序,旨在增强DPA-1模型对钙钛矿型氧化物的描述能力.力场开发程序的工作流程如图6b[107]所示,该方法旨在获得通用的钙钛矿型氧化物原子间力场.初始数据集包含约1000个结构,涵盖26种不同类型的钙钛矿氧化物,涉及约200个组分和14种金属元素.首先,使用标准的DP-GEN策略来收敛3种元素的钙钛矿氧化物的DPA-1模型.然后,将收敛的训练数据集作为DP-GEN的起点,以获得适用于4种元素钙钛矿体系的DPA-1模型,例如PbZr1 - x Ti x O3和Pb(Mg1/3Nb2/3)O3.最终得到一个能够描述包括Pb(In1/2Nb1/2)O3-Pb(Mg1/3Nb2/3)O3-PbTiO3 (即PIN-PMN-PT)等三元固溶体在内的6种元素的钙钛矿型氧化物的DPA-1模型,该模型即为钙钛矿型铁电材料通用势场,命名为UniPero,能够描述多种钙钛矿型氧化物. ...
... [107]所示,该方法旨在获得通用的钙钛矿型氧化物原子间力场.初始数据集包含约1000个结构,涵盖26种不同类型的钙钛矿氧化物,涉及约200个组分和14种金属元素.首先,使用标准的DP-GEN策略来收敛3种元素的钙钛矿氧化物的DPA-1模型.然后,将收敛的训练数据集作为DP-GEN的起点,以获得适用于4种元素钙钛矿体系的DPA-1模型,例如PbZr1 - x Ti x O3和Pb(Mg1/3Nb2/3)O3.最终得到一个能够描述包括Pb(In1/2Nb1/2)O3-Pb(Mg1/3Nb2/3)O3-PbTiO3 (即PIN-PMN-PT)等三元固溶体在内的6种元素的钙钛矿型氧化物的DPA-1模型,该模型即为钙钛矿型铁电材料通用势场,命名为UniPero,能够描述多种钙钛矿型氧化物. ...
... 在确认模型能量和力准确收敛的前提下,DPA-1模型适用性可通过模拟钙钛矿型铁电材料的温度驱动相变来进行测试.如图7[107]所示,DPA-1模型在没有任何调整的情况下,成功地复现了铅基铁电体(如PbTiO3、Pb0.5Sr0.5TiO3和PbZr0.5Ti0.5O3)、无铅铁电体(如BaTiO3、KNbO3和K0.5Na0.5NbO3)、量子顺电体SrTiO3以及三元固溶体(0.29PIN-0.45PMN-0.26PT和0.36PIN-0.36PMN-0.28PT)温度驱动的相变过程,模拟结果与实验结果具有很好的一致性.基于NPT系综的分子动力学模拟得到温度与晶格常数的关系显示:在PbTiO3中,模拟预测了从四方晶系(T,空间群P4mm)向立方晶系(C,空间群Pmm)的相转变,相变温度(Tc)约为550 K (图7a[107]),而在A位掺杂的Pb0.5Sr0.5TiO3固溶体中,Tc降至约280 K (图7d[107]).相比之下,使用DPA-1模型模拟的B位掺杂的PbZr0.5Ti0.5O3固溶体,在低温下为菱形结构(R,空间群R3m),随着温度的升高发生了菱形-四方-立方(R-T-C)的相变(图7g[107]),与实验结果一致.量子顺电体SrTiO3随着温度的升高表现出四方-立方相变(I4/mcm→Pmm),其特征是TiO6八面体倾斜角的减小.这个相变对应着一个约为1 meV/atom的微小能垒,DPA-1模型成功地复现了该相变过程(图7b[107]),理论上的Tc值为150 K,与实验值105 K相当.此外,DPA-1模型还正确预测了BaTiO3和K0.5Na0.5NbO3中的相变过程,即菱形-正交-四方-立方(R-O-T-C) (图7c和h[107]).对于像PIN-PMN-PT这样复杂的包含6种元素的固溶体,DPA-1不仅能够复现实验中观察到的T-C相变,还能够正确预测由组分变化导致的Tc的变化趋势.如图7f和i[107]所示,0.29PIN-0.45PMN-0.26PT到0.36PIN-0.36PMN-0.28PT相变温度约升高50 K,类似于实验中PIN的组分含量从29%增加到36%,而Tc相应升高约60 K.值得说明的是,铁电相变的理论Tc值通常比实验值低100~200 K.这种差异是通过拟合PBEsol密度泛函计算结果所开发的力场的常见特征,考虑到DPA-1模型预测结果与DFT计算结果表现出的高度的一致性,Tc的低估更可能是由于PBEsol在预测有限温度方面具有局限性,而不是DPA-1模型精度的问题. ...
... [107]Correlation of temperature with lattice constant based on generalized interatomic potential simulations (T—tetragonal, C—cubic, R—rhombohedral, O—orthorhombic)
... [116]Schematic representation of the inverse ferroelectric distortion octahedral turning angle (<i>φ</i>) (<i>φ<sub>n </sub></i> is defined as the antiferrodistortive (AFD) order parameter, where the index <i>n</i> is the sequence number of unit cell, and <i>θ</i> is the rotation angle of TiO<sub>6</sub> octahedra in each unit cell) (a), phase diagram of the SrTiO<sub>3</sub> (STO) block under biaxial in-plane strain simulated by deep potential molecular dynamics (The ferroelectric (FE) and AFD transition temperatures at different strains are indicated by blue square dots and red dots, respectively. <i>T</i> represents temperature) (b), and curves of polarization and octahedral turning angle (<i>ϕ</i>) versus temperature for STO supercells (5000 atoms) under biaxial in-plane strain (-0.8%) (c)<sup>[<xref ref-type="bibr" rid="R116">116</xref>]</sup>Fig.8
... [117]Schematic of the domain lattice point model in SrTiO<sub>3</sub> (STO) films (Where the domains generate spontaneous concave and convex undulations in the (001) plane, with the convex angle corresponding to the domains with upward polarization and the concave angle corresponding to the domains with downward polarization, and the domains can be oriented to rotate in order to flip the polarization. <i>a</i> and <i>c</i> are lattice parameters, and <i>α</i> is the angel between <i>a-c</i> domain) (a), schematic diagram of a 90° rotation of polarized domain boundaries by bending deformation (<i>P</i><sub>macro</sub> is the macroscopic polarization, head-to-tail and head-to-head domain configurations are denoted as HT and HH, respectively) (b), and hysteresis-like features of the STO film under strain, and after the stress is removed, the STO can still maintain the bent state with residual polarization (c)<sup>[<xref ref-type="bibr" rid="R117">117</xref>]</sup>Fig.9<strong>4.2 HfO<sub>2</sub></strong> 高<strong>O<sup>2</sup></strong><sup>-</sup>迁移率的动力学研究
... [128]Polarization reversal paths in the ferroelectric phase HfO<sub>2</sub><sup> [<xref ref-type="bibr" rid="R128">128</xref>]</sup>
(a) vibrational modes of crystals in a single cell of Pca21, with outwardly and inwardly shifted oxygen atoms indicated by purple and brown-orange spheres, respectively, and polarization along the z-axis ...
... [128]
(a) vibrational modes of crystals in a single cell of Pca21, with outwardly and inwardly shifted oxygen atoms indicated by purple and brown-orange spheres, respectively, and polarization along the z-axis ...
... [128]Oxygen ion transport is coupled to the nucleation and growth mechanism of the ferroelectric polarization reversal<sup>[<xref ref-type="bibr" rid="R128">128</xref>]</sup>
(a) polar-antipolar phase cycling induced by successive SI and SA ferroelectric transitions. The highlighted marker Onp in the initial structure, δ = δ0 (defined as displacement of the Op ion relative to the top Hf plane), becomes Op after SI-2, and δ = -δ0, becomes Op after SA ...
... [128]
(a) polar-antipolar phase cycling induced by successive SI and SA ferroelectric transitions. The highlighted marker Onp in the initial structure, δ = δ0 (defined as displacement of the Op ion relative to the top Hf plane), becomes Op after SI-2, and δ = -δ0, becomes Op after SA ...








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}