Thermodynamic tuning of Mg-based hydrogen storage alloys: A review
2
2013
... 能源是发展现代工业文明和实现人类可持续发展的核心动力.随着全球对“碳达峰”和“碳中和”达成共识,开发清洁、高效的可再生能源并建立健全相关的能源生产供应体系势在必行.作为一种高能量密度(142 MJ/kg)的清洁能源,氢能在未来能源系统中将扮演重要角色.目前,H2的安全、高密度可逆存储仍制约着氢能规模化应用,是亟待解决的关键技术.目前研究开发的储氢技术主要有固态储氢、高压气态储氢、有机液体储氢和低温液态储氢等4种方式.其中,高压气态储氢技术已在燃料电池汽车实现商业应用,但体积储氢密度仍较低,而高达35甚至70 MPa的储氢压力,对储氢罐的制造提出了极高的要求,且高压氢的安全性令人担忧,加氢站建设成本高.有机液体储氢(LOHC)技术具有储氢密度高、存储设备简单等特点,特别是以咔唑和甲酸等为代表的液态有机体系能够在较低温度下脱氢.但液态有机储氢技术同样存在一些缺点,例如加氢温度过高、脱氢效率较低、催化剂失效以及过程中容易发生副反应等.此外,低温液氢储氢技术虽然质量和体积储氢密度都较高,但H2的液化过程却消耗了所储存H2总能量的30%~45%,在运输和保存过程中对低温容器也要求极高,大规模应用仍有很大的局限性[1,2].固态储氢具有高体积密度、高安全、装备和基础设施易实现等突出优势,但其质量储氢密度仍偏低且材料成本较高[3].因此,开发低成本、高质量储氢密度的固态储氢材料对于储氢技术发展具有重要意义. ...
... 金属氢化物的吸/放氢反应是典型的气-固反应,脱氢反应是吸氢反应的逆过程.纯Mg的氢化过程主要包括[6]:① H2分子在Mg表面吸附、解离为H原子;② H原子在Mg表面渗透并向内部扩散;③ 形成MgHx固溶体;④ 固溶度达到一定程度时,发生相变生成MgH2.对于金属Mg的氢化过程,除了表面生成的MgO会阻碍反应动力学外,一般认为其限制反应动力学的主要因素包括H2在纯Mg表面较低的解离速率和H原子在反应生成的MgH2中较低的扩散速率[7].另外,氢化反应过程中H2压力对反应动力学有一定影响[8],实际上,影响该反应进行的原因更为复杂.例如,Ouyang等[9]通过控制磁控溅射的条件制备了密排面(0001)织构和非密排面()织构的Mg薄膜,发现具有()取向的Mg薄膜的吸氢温度要比(0001)取向的Mg薄膜低200℃,总的吸氢量达到了5.6%.理论计算表明,()面对于吸氢反应中的氢渗透过程阻碍较小,H在迁移过程中所需能垒较低,进而有利于吸氢反应的进行.虽然Mg/MgH2吸/放氢反应速率的限制因素尚有待进一步探索,但研究[1]表明,添加催化剂、纳米化/纳米限域(将储氢材料装载到纳米尺寸的空间内)以及多相复合等手段可以显著改善MgH2的吸/放氢动力学.下文将主要从添加催化剂、纳米化/纳米限域以及多相复合等方面阐述镁基储氢合金在吸/放氢反应动力学调控方面取得的进展. ...
Current research progress and perspectives on liquid hydrogen rich molecules in sustainable hydrogen storage
1
2021
... 能源是发展现代工业文明和实现人类可持续发展的核心动力.随着全球对“碳达峰”和“碳中和”达成共识,开发清洁、高效的可再生能源并建立健全相关的能源生产供应体系势在必行.作为一种高能量密度(142 MJ/kg)的清洁能源,氢能在未来能源系统中将扮演重要角色.目前,H2的安全、高密度可逆存储仍制约着氢能规模化应用,是亟待解决的关键技术.目前研究开发的储氢技术主要有固态储氢、高压气态储氢、有机液体储氢和低温液态储氢等4种方式.其中,高压气态储氢技术已在燃料电池汽车实现商业应用,但体积储氢密度仍较低,而高达35甚至70 MPa的储氢压力,对储氢罐的制造提出了极高的要求,且高压氢的安全性令人担忧,加氢站建设成本高.有机液体储氢(LOHC)技术具有储氢密度高、存储设备简单等特点,特别是以咔唑和甲酸等为代表的液态有机体系能够在较低温度下脱氢.但液态有机储氢技术同样存在一些缺点,例如加氢温度过高、脱氢效率较低、催化剂失效以及过程中容易发生副反应等.此外,低温液氢储氢技术虽然质量和体积储氢密度都较高,但H2的液化过程却消耗了所储存H2总能量的30%~45%,在运输和保存过程中对低温容器也要求极高,大规模应用仍有很大的局限性[1,2].固态储氢具有高体积密度、高安全、装备和基础设施易实现等突出优势,但其质量储氢密度仍偏低且材料成本较高[3].因此,开发低成本、高质量储氢密度的固态储氢材料对于储氢技术发展具有重要意义. ...
Mg-based materials for hydrogen storage
1
2021
... 能源是发展现代工业文明和实现人类可持续发展的核心动力.随着全球对“碳达峰”和“碳中和”达成共识,开发清洁、高效的可再生能源并建立健全相关的能源生产供应体系势在必行.作为一种高能量密度(142 MJ/kg)的清洁能源,氢能在未来能源系统中将扮演重要角色.目前,H2的安全、高密度可逆存储仍制约着氢能规模化应用,是亟待解决的关键技术.目前研究开发的储氢技术主要有固态储氢、高压气态储氢、有机液体储氢和低温液态储氢等4种方式.其中,高压气态储氢技术已在燃料电池汽车实现商业应用,但体积储氢密度仍较低,而高达35甚至70 MPa的储氢压力,对储氢罐的制造提出了极高的要求,且高压氢的安全性令人担忧,加氢站建设成本高.有机液体储氢(LOHC)技术具有储氢密度高、存储设备简单等特点,特别是以咔唑和甲酸等为代表的液态有机体系能够在较低温度下脱氢.但液态有机储氢技术同样存在一些缺点,例如加氢温度过高、脱氢效率较低、催化剂失效以及过程中容易发生副反应等.此外,低温液氢储氢技术虽然质量和体积储氢密度都较高,但H2的液化过程却消耗了所储存H2总能量的30%~45%,在运输和保存过程中对低温容器也要求极高,大规模应用仍有很大的局限性[1,2].固态储氢具有高体积密度、高安全、装备和基础设施易实现等突出优势,但其质量储氢密度仍偏低且材料成本较高[3].因此,开发低成本、高质量储氢密度的固态储氢材料对于储氢技术发展具有重要意义. ...
Empowering hydrogen storage performance of MgH2 by nanoengineering and nanocatalysis
1
2020
... 一般而言,根据H2与材料的相互作用,可将储氢材料分为物理吸附材料与氢化物储氢材料两大类.本文讨论的材料属于后者.在氢化物储氢材料中,早期的研究主要集中于AB2、AB5型等合金上,其质量储氢密度大多小于2% (质量分数,下同),低于车载储氢应用的标准,且富含较贵的稀土、Ni、Zr等元素,成本较高.轻金属氢化物MgH2质量储氢密度高达7.6%,资源丰富且成本相对较低,是十分有发展潜力的储氢材料.但由于其热力学过于稳定,脱氢反应焓变高达76 kJ/mol[4],导致工作温度往往超过300℃.而长时间高温工作会带来材料组织结构变化、循环性能下降的问题.同时,过高的放氢温度难以匹配燃料电池工作模块温度(80℃).此外,MgH2缓慢的吸/放氢反应动力学也难以满足储氢装置加氢、供氢速率的实际需求.另一方面,虽然Ni-MH电池在能量密度、循环寿命上不及锂离子电池,但其在环境友好性,尤其是在宽温区工作、安全性方面拥有独特优势.因此,目前在电动工具、智能家电、汽车紧急呼救系统、油电混合动力汽车领域仍大量使用.但是进一步提高其电化学容量,仍是这类负极材料面临的艰难挑战.自Kadir等[5]发现AB3型稀土(RE)-Mg-Ni合金的电化学容量显著高于广泛使用的AB5混合稀土合金以来,这类合金及随后衍生出的A2B7型RE-Mg-Ni合金展现出良好的前景,并逐步在Ni-MH电池负极材料上得到应用.此外,Mg-Ni基非晶储氢合金负极材料具有更高的电化学容量.因此,镁基储氢合金在电化学储能方面也有重要地位. ...
Structural investigation and hydrogen storage capacity of LaMg2Ni9 and (La0.65Ca0.35) (Mg1.32Ca0.68) Ni9 of the AB2C9 type structure
1
2000
... 一般而言,根据H2与材料的相互作用,可将储氢材料分为物理吸附材料与氢化物储氢材料两大类.本文讨论的材料属于后者.在氢化物储氢材料中,早期的研究主要集中于AB2、AB5型等合金上,其质量储氢密度大多小于2% (质量分数,下同),低于车载储氢应用的标准,且富含较贵的稀土、Ni、Zr等元素,成本较高.轻金属氢化物MgH2质量储氢密度高达7.6%,资源丰富且成本相对较低,是十分有发展潜力的储氢材料.但由于其热力学过于稳定,脱氢反应焓变高达76 kJ/mol[4],导致工作温度往往超过300℃.而长时间高温工作会带来材料组织结构变化、循环性能下降的问题.同时,过高的放氢温度难以匹配燃料电池工作模块温度(80℃).此外,MgH2缓慢的吸/放氢反应动力学也难以满足储氢装置加氢、供氢速率的实际需求.另一方面,虽然Ni-MH电池在能量密度、循环寿命上不及锂离子电池,但其在环境友好性,尤其是在宽温区工作、安全性方面拥有独特优势.因此,目前在电动工具、智能家电、汽车紧急呼救系统、油电混合动力汽车领域仍大量使用.但是进一步提高其电化学容量,仍是这类负极材料面临的艰难挑战.自Kadir等[5]发现AB3型稀土(RE)-Mg-Ni合金的电化学容量显著高于广泛使用的AB5混合稀土合金以来,这类合金及随后衍生出的A2B7型RE-Mg-Ni合金展现出良好的前景,并逐步在Ni-MH电池负极材料上得到应用.此外,Mg-Ni基非晶储氢合金负极材料具有更高的电化学容量.因此,镁基储氢合金在电化学储能方面也有重要地位. ...
Dehydriding kinetics of a mechanically alloyed mixture Mg-10wt. %Ni
1
1999
... 金属氢化物的吸/放氢反应是典型的气-固反应,脱氢反应是吸氢反应的逆过程.纯Mg的氢化过程主要包括[6]:① H2分子在Mg表面吸附、解离为H原子;② H原子在Mg表面渗透并向内部扩散;③ 形成MgHx固溶体;④ 固溶度达到一定程度时,发生相变生成MgH2.对于金属Mg的氢化过程,除了表面生成的MgO会阻碍反应动力学外,一般认为其限制反应动力学的主要因素包括H2在纯Mg表面较低的解离速率和H原子在反应生成的MgH2中较低的扩散速率[7].另外,氢化反应过程中H2压力对反应动力学有一定影响[8],实际上,影响该反应进行的原因更为复杂.例如,Ouyang等[9]通过控制磁控溅射的条件制备了密排面(0001)织构和非密排面()织构的Mg薄膜,发现具有()取向的Mg薄膜的吸氢温度要比(0001)取向的Mg薄膜低200℃,总的吸氢量达到了5.6%.理论计算表明,()面对于吸氢反应中的氢渗透过程阻碍较小,H在迁移过程中所需能垒较低,进而有利于吸氢反应的进行.虽然Mg/MgH2吸/放氢反应速率的限制因素尚有待进一步探索,但研究[1]表明,添加催化剂、纳米化/纳米限域(将储氢材料装载到纳米尺寸的空间内)以及多相复合等手段可以显著改善MgH2的吸/放氢动力学.下文将主要从添加催化剂、纳米化/纳米限域以及多相复合等方面阐述镁基储氢合金在吸/放氢反应动力学调控方面取得的进展. ...
Hydrogen in magnesium: New perspectives toward functional stores
1
2010
... 金属氢化物的吸/放氢反应是典型的气-固反应,脱氢反应是吸氢反应的逆过程.纯Mg的氢化过程主要包括[6]:① H2分子在Mg表面吸附、解离为H原子;② H原子在Mg表面渗透并向内部扩散;③ 形成MgHx固溶体;④ 固溶度达到一定程度时,发生相变生成MgH2.对于金属Mg的氢化过程,除了表面生成的MgO会阻碍反应动力学外,一般认为其限制反应动力学的主要因素包括H2在纯Mg表面较低的解离速率和H原子在反应生成的MgH2中较低的扩散速率[7].另外,氢化反应过程中H2压力对反应动力学有一定影响[8],实际上,影响该反应进行的原因更为复杂.例如,Ouyang等[9]通过控制磁控溅射的条件制备了密排面(0001)织构和非密排面()织构的Mg薄膜,发现具有()取向的Mg薄膜的吸氢温度要比(0001)取向的Mg薄膜低200℃,总的吸氢量达到了5.6%.理论计算表明,()面对于吸氢反应中的氢渗透过程阻碍较小,H在迁移过程中所需能垒较低,进而有利于吸氢反应的进行.虽然Mg/MgH2吸/放氢反应速率的限制因素尚有待进一步探索,但研究[1]表明,添加催化剂、纳米化/纳米限域(将储氢材料装载到纳米尺寸的空间内)以及多相复合等手段可以显著改善MgH2的吸/放氢动力学.下文将主要从添加催化剂、纳米化/纳米限域以及多相复合等方面阐述镁基储氢合金在吸/放氢反应动力学调控方面取得的进展. ...
Improving the hydrogenation properties of MgH2 at room temperature by doping with nano-size ZrO2 catalyst
1
2016
... 金属氢化物的吸/放氢反应是典型的气-固反应,脱氢反应是吸氢反应的逆过程.纯Mg的氢化过程主要包括[6]:① H2分子在Mg表面吸附、解离为H原子;② H原子在Mg表面渗透并向内部扩散;③ 形成MgHx固溶体;④ 固溶度达到一定程度时,发生相变生成MgH2.对于金属Mg的氢化过程,除了表面生成的MgO会阻碍反应动力学外,一般认为其限制反应动力学的主要因素包括H2在纯Mg表面较低的解离速率和H原子在反应生成的MgH2中较低的扩散速率[7].另外,氢化反应过程中H2压力对反应动力学有一定影响[8],实际上,影响该反应进行的原因更为复杂.例如,Ouyang等[9]通过控制磁控溅射的条件制备了密排面(0001)织构和非密排面()织构的Mg薄膜,发现具有()取向的Mg薄膜的吸氢温度要比(0001)取向的Mg薄膜低200℃,总的吸氢量达到了5.6%.理论计算表明,()面对于吸氢反应中的氢渗透过程阻碍较小,H在迁移过程中所需能垒较低,进而有利于吸氢反应的进行.虽然Mg/MgH2吸/放氢反应速率的限制因素尚有待进一步探索,但研究[1]表明,添加催化剂、纳米化/纳米限域(将储氢材料装载到纳米尺寸的空间内)以及多相复合等手段可以显著改善MgH2的吸/放氢动力学.下文将主要从添加催化剂、纳米化/纳米限域以及多相复合等方面阐述镁基储氢合金在吸/放氢反应动力学调控方面取得的进展. ...
Express penetration of hydrogen on Mg() along the close-packed-planes
1
2015
... 金属氢化物的吸/放氢反应是典型的气-固反应,脱氢反应是吸氢反应的逆过程.纯Mg的氢化过程主要包括[6]:① H2分子在Mg表面吸附、解离为H原子;② H原子在Mg表面渗透并向内部扩散;③ 形成MgHx固溶体;④ 固溶度达到一定程度时,发生相变生成MgH2.对于金属Mg的氢化过程,除了表面生成的MgO会阻碍反应动力学外,一般认为其限制反应动力学的主要因素包括H2在纯Mg表面较低的解离速率和H原子在反应生成的MgH2中较低的扩散速率[7].另外,氢化反应过程中H2压力对反应动力学有一定影响[8],实际上,影响该反应进行的原因更为复杂.例如,Ouyang等[9]通过控制磁控溅射的条件制备了密排面(0001)织构和非密排面()织构的Mg薄膜,发现具有()取向的Mg薄膜的吸氢温度要比(0001)取向的Mg薄膜低200℃,总的吸氢量达到了5.6%.理论计算表明,()面对于吸氢反应中的氢渗透过程阻碍较小,H在迁移过程中所需能垒较低,进而有利于吸氢反应的进行.虽然Mg/MgH2吸/放氢反应速率的限制因素尚有待进一步探索,但研究[1]表明,添加催化剂、纳米化/纳米限域(将储氢材料装载到纳米尺寸的空间内)以及多相复合等手段可以显著改善MgH2的吸/放氢动力学.下文将主要从添加催化剂、纳米化/纳米限域以及多相复合等方面阐述镁基储氢合金在吸/放氢反应动力学调控方面取得的进展. ...
Kinetics in Mg-based hydrogen storage materials: Enhancement and mechanism
1
2019
... MgH2的脱氢动力学速率缓慢,脱氢活化能高达160 kJ/mol,使用催化剂可加速氢分子在Mg/MgH2表面的解离和重构,从而有效降低脱氢反应的活化能[10].研究表明,添加Ti[11,12]、Nb[13]、V[14]等具有多价态的过渡金属及其化合物有突出的催化效果,能够显著降低脱氢反应的活化能.例如:Zhang等[15]将合成的多价态(0~+5)非晶NbHx加入到MgH2球磨,发现球磨后的复合材料可以在300℃、3 kPa的初始氢压下9 min内脱出7%的H2,认为纳米级的NbHx提供了H快速扩散的途径,而多价态的铌离子是反应过程中电子转移的良好媒介,因而产生了良好的催化效果.Wang等[16]将N掺杂的Nb2O5加入到MgH2中球磨,获得的Mg + 10%N-Nb2O5复合材料在120℃、50 Pa氢压下3 min内吸氢5.1%,且在170℃开始脱氢,250℃可脱氢5.0%.密度泛函理论(DFT)计算表明Nb-N-O活性物质弱化了Mg-H键,从而显著降低MgH2的脱氢温度,并且吸/放氢循环过程中形成的稳定Nb-N-O活性物质加速了氢分子的解离和重构.同样,不同价态的CeF3/CeF4添加对MgH2的储氢性能也有不同程度的改善[17],例如MgH2 + CeF3复合材料中CeF3对MgH2脱氢反应的催化效果不明显,而MgH2 + CeF4复合材料的脱氢峰的峰值温度要比MgH2低40℃,这可能是由于高价态的Ce4+更容易导致反应电子的转移. ...
Superior catalytic activity derived from a two-dimensional Ti3C2 precursor towards the hydrogen storage reaction of magnesium hydride
1
2016
... MgH2的脱氢动力学速率缓慢,脱氢活化能高达160 kJ/mol,使用催化剂可加速氢分子在Mg/MgH2表面的解离和重构,从而有效降低脱氢反应的活化能[10].研究表明,添加Ti[11,12]、Nb[13]、V[14]等具有多价态的过渡金属及其化合物有突出的催化效果,能够显著降低脱氢反应的活化能.例如:Zhang等[15]将合成的多价态(0~+5)非晶NbHx加入到MgH2球磨,发现球磨后的复合材料可以在300℃、3 kPa的初始氢压下9 min内脱出7%的H2,认为纳米级的NbHx提供了H快速扩散的途径,而多价态的铌离子是反应过程中电子转移的良好媒介,因而产生了良好的催化效果.Wang等[16]将N掺杂的Nb2O5加入到MgH2中球磨,获得的Mg + 10%N-Nb2O5复合材料在120℃、50 Pa氢压下3 min内吸氢5.1%,且在170℃开始脱氢,250℃可脱氢5.0%.密度泛函理论(DFT)计算表明Nb-N-O活性物质弱化了Mg-H键,从而显著降低MgH2的脱氢温度,并且吸/放氢循环过程中形成的稳定Nb-N-O活性物质加速了氢分子的解离和重构.同样,不同价态的CeF3/CeF4添加对MgH2的储氢性能也有不同程度的改善[17],例如MgH2 + CeF3复合材料中CeF3对MgH2脱氢反应的催化效果不明显,而MgH2 + CeF4复合材料的脱氢峰的峰值温度要比MgH2低40℃,这可能是由于高价态的Ce4+更容易导致反应电子的转移. ...
Enhanced hydrogen storage properties of MgH2 catalyzed with carbon-supported nanocrystalline TiO2
1
2018
... MgH2的脱氢动力学速率缓慢,脱氢活化能高达160 kJ/mol,使用催化剂可加速氢分子在Mg/MgH2表面的解离和重构,从而有效降低脱氢反应的活化能[10].研究表明,添加Ti[11,12]、Nb[13]、V[14]等具有多价态的过渡金属及其化合物有突出的催化效果,能够显著降低脱氢反应的活化能.例如:Zhang等[15]将合成的多价态(0~+5)非晶NbHx加入到MgH2球磨,发现球磨后的复合材料可以在300℃、3 kPa的初始氢压下9 min内脱出7%的H2,认为纳米级的NbHx提供了H快速扩散的途径,而多价态的铌离子是反应过程中电子转移的良好媒介,因而产生了良好的催化效果.Wang等[16]将N掺杂的Nb2O5加入到MgH2中球磨,获得的Mg + 10%N-Nb2O5复合材料在120℃、50 Pa氢压下3 min内吸氢5.1%,且在170℃开始脱氢,250℃可脱氢5.0%.密度泛函理论(DFT)计算表明Nb-N-O活性物质弱化了Mg-H键,从而显著降低MgH2的脱氢温度,并且吸/放氢循环过程中形成的稳定Nb-N-O活性物质加速了氢分子的解离和重构.同样,不同价态的CeF3/CeF4添加对MgH2的储氢性能也有不同程度的改善[17],例如MgH2 + CeF3复合材料中CeF3对MgH2脱氢反应的催化效果不明显,而MgH2 + CeF4复合材料的脱氢峰的峰值温度要比MgH2低40℃,这可能是由于高价态的Ce4+更容易导致反应电子的转移. ...
Graphene-induced growth of N-doped niobium pentaoxide nanorods with high catalytic activity for hydrogen storage in MgH2
1
2021
... MgH2的脱氢动力学速率缓慢,脱氢活化能高达160 kJ/mol,使用催化剂可加速氢分子在Mg/MgH2表面的解离和重构,从而有效降低脱氢反应的活化能[10].研究表明,添加Ti[11,12]、Nb[13]、V[14]等具有多价态的过渡金属及其化合物有突出的催化效果,能够显著降低脱氢反应的活化能.例如:Zhang等[15]将合成的多价态(0~+5)非晶NbHx加入到MgH2球磨,发现球磨后的复合材料可以在300℃、3 kPa的初始氢压下9 min内脱出7%的H2,认为纳米级的NbHx提供了H快速扩散的途径,而多价态的铌离子是反应过程中电子转移的良好媒介,因而产生了良好的催化效果.Wang等[16]将N掺杂的Nb2O5加入到MgH2中球磨,获得的Mg + 10%N-Nb2O5复合材料在120℃、50 Pa氢压下3 min内吸氢5.1%,且在170℃开始脱氢,250℃可脱氢5.0%.密度泛函理论(DFT)计算表明Nb-N-O活性物质弱化了Mg-H键,从而显著降低MgH2的脱氢温度,并且吸/放氢循环过程中形成的稳定Nb-N-O活性物质加速了氢分子的解离和重构.同样,不同价态的CeF3/CeF4添加对MgH2的储氢性能也有不同程度的改善[17],例如MgH2 + CeF3复合材料中CeF3对MgH2脱氢反应的催化效果不明显,而MgH2 + CeF4复合材料的脱氢峰的峰值温度要比MgH2低40℃,这可能是由于高价态的Ce4+更容易导致反应电子的转移. ...
Vanadium oxide nanoparticles supported on cubic carbon nanoboxes as highly active catalyst precursors for hydrogen storage in MgH2
1
2018
... MgH2的脱氢动力学速率缓慢,脱氢活化能高达160 kJ/mol,使用催化剂可加速氢分子在Mg/MgH2表面的解离和重构,从而有效降低脱氢反应的活化能[10].研究表明,添加Ti[11,12]、Nb[13]、V[14]等具有多价态的过渡金属及其化合物有突出的催化效果,能够显著降低脱氢反应的活化能.例如:Zhang等[15]将合成的多价态(0~+5)非晶NbHx加入到MgH2球磨,发现球磨后的复合材料可以在300℃、3 kPa的初始氢压下9 min内脱出7%的H2,认为纳米级的NbHx提供了H快速扩散的途径,而多价态的铌离子是反应过程中电子转移的良好媒介,因而产生了良好的催化效果.Wang等[16]将N掺杂的Nb2O5加入到MgH2中球磨,获得的Mg + 10%N-Nb2O5复合材料在120℃、50 Pa氢压下3 min内吸氢5.1%,且在170℃开始脱氢,250℃可脱氢5.0%.密度泛函理论(DFT)计算表明Nb-N-O活性物质弱化了Mg-H键,从而显著降低MgH2的脱氢温度,并且吸/放氢循环过程中形成的稳定Nb-N-O活性物质加速了氢分子的解离和重构.同样,不同价态的CeF3/CeF4添加对MgH2的储氢性能也有不同程度的改善[17],例如MgH2 + CeF3复合材料中CeF3对MgH2脱氢反应的催化效果不明显,而MgH2 + CeF4复合材料的脱氢峰的峰值温度要比MgH2低40℃,这可能是由于高价态的Ce4+更容易导致反应电子的转移. ...
Remarkably improved hydrogen storage performance of MgH2 catalyzed by multivalence NbHx nanoparticles
1
2015
... MgH2的脱氢动力学速率缓慢,脱氢活化能高达160 kJ/mol,使用催化剂可加速氢分子在Mg/MgH2表面的解离和重构,从而有效降低脱氢反应的活化能[10].研究表明,添加Ti[11,12]、Nb[13]、V[14]等具有多价态的过渡金属及其化合物有突出的催化效果,能够显著降低脱氢反应的活化能.例如:Zhang等[15]将合成的多价态(0~+5)非晶NbHx加入到MgH2球磨,发现球磨后的复合材料可以在300℃、3 kPa的初始氢压下9 min内脱出7%的H2,认为纳米级的NbHx提供了H快速扩散的途径,而多价态的铌离子是反应过程中电子转移的良好媒介,因而产生了良好的催化效果.Wang等[16]将N掺杂的Nb2O5加入到MgH2中球磨,获得的Mg + 10%N-Nb2O5复合材料在120℃、50 Pa氢压下3 min内吸氢5.1%,且在170℃开始脱氢,250℃可脱氢5.0%.密度泛函理论(DFT)计算表明Nb-N-O活性物质弱化了Mg-H键,从而显著降低MgH2的脱氢温度,并且吸/放氢循环过程中形成的稳定Nb-N-O活性物质加速了氢分子的解离和重构.同样,不同价态的CeF3/CeF4添加对MgH2的储氢性能也有不同程度的改善[17],例如MgH2 + CeF3复合材料中CeF3对MgH2脱氢反应的催化效果不明显,而MgH2 + CeF4复合材料的脱氢峰的峰值温度要比MgH2低40℃,这可能是由于高价态的Ce4+更容易导致反应电子的转移. ...
Nitrogen-stimulated superior catalytic activity of niobium oxide for fast full hydrogenation of magnesium at ambient temperature
1
2019
... MgH2的脱氢动力学速率缓慢,脱氢活化能高达160 kJ/mol,使用催化剂可加速氢分子在Mg/MgH2表面的解离和重构,从而有效降低脱氢反应的活化能[10].研究表明,添加Ti[11,12]、Nb[13]、V[14]等具有多价态的过渡金属及其化合物有突出的催化效果,能够显著降低脱氢反应的活化能.例如:Zhang等[15]将合成的多价态(0~+5)非晶NbHx加入到MgH2球磨,发现球磨后的复合材料可以在300℃、3 kPa的初始氢压下9 min内脱出7%的H2,认为纳米级的NbHx提供了H快速扩散的途径,而多价态的铌离子是反应过程中电子转移的良好媒介,因而产生了良好的催化效果.Wang等[16]将N掺杂的Nb2O5加入到MgH2中球磨,获得的Mg + 10%N-Nb2O5复合材料在120℃、50 Pa氢压下3 min内吸氢5.1%,且在170℃开始脱氢,250℃可脱氢5.0%.密度泛函理论(DFT)计算表明Nb-N-O活性物质弱化了Mg-H键,从而显著降低MgH2的脱氢温度,并且吸/放氢循环过程中形成的稳定Nb-N-O活性物质加速了氢分子的解离和重构.同样,不同价态的CeF3/CeF4添加对MgH2的储氢性能也有不同程度的改善[17],例如MgH2 + CeF3复合材料中CeF3对MgH2脱氢反应的催化效果不明显,而MgH2 + CeF4复合材料的脱氢峰的峰值温度要比MgH2低40℃,这可能是由于高价态的Ce4+更容易导致反应电子的转移. ...
Enhanced hydrogen desorption property of MgH2 with the addition of cerium fluorides
1
2015
... MgH2的脱氢动力学速率缓慢,脱氢活化能高达160 kJ/mol,使用催化剂可加速氢分子在Mg/MgH2表面的解离和重构,从而有效降低脱氢反应的活化能[10].研究表明,添加Ti[11,12]、Nb[13]、V[14]等具有多价态的过渡金属及其化合物有突出的催化效果,能够显著降低脱氢反应的活化能.例如:Zhang等[15]将合成的多价态(0~+5)非晶NbHx加入到MgH2球磨,发现球磨后的复合材料可以在300℃、3 kPa的初始氢压下9 min内脱出7%的H2,认为纳米级的NbHx提供了H快速扩散的途径,而多价态的铌离子是反应过程中电子转移的良好媒介,因而产生了良好的催化效果.Wang等[16]将N掺杂的Nb2O5加入到MgH2中球磨,获得的Mg + 10%N-Nb2O5复合材料在120℃、50 Pa氢压下3 min内吸氢5.1%,且在170℃开始脱氢,250℃可脱氢5.0%.密度泛函理论(DFT)计算表明Nb-N-O活性物质弱化了Mg-H键,从而显著降低MgH2的脱氢温度,并且吸/放氢循环过程中形成的稳定Nb-N-O活性物质加速了氢分子的解离和重构.同样,不同价态的CeF3/CeF4添加对MgH2的储氢性能也有不同程度的改善[17],例如MgH2 + CeF3复合材料中CeF3对MgH2脱氢反应的催化效果不明显,而MgH2 + CeF4复合材料的脱氢峰的峰值温度要比MgH2低40℃,这可能是由于高价态的Ce4+更容易导致反应电子的转移. ...
Remarkable enhancement in dehydrogenation of MgH2 by a nano-coating of multi-valence Ti-based catalysts
5
2013
... 为进一步揭示多价态过渡金属及其化合物的催化机理,并获得更好的催化效果,Cui等[18]将球磨预处理后的Mg粉与过渡金属氯化物TiCl3在四氢呋喃(C4H8O)中反应制备了多相多价态钛基催化剂包覆的Mg颗粒,其脱氢活化能降至30.8 kJ/mol,起始脱氢温度约为175℃,比球磨MgH2、Mg-TiCl3材料分别降低了105和60℃,如图1a[18]所示.这是由于多相多价态的钛基催化剂可以作为Mg/MgH2吸/放氢过程中电子转移的载体,如图1b[18]所示,通过多价态的钛催化完成吸/放氢反应过程中的电子转移要比Mg和H或Mg2+和H-间直接转移电子更加容易,从而降低了反应活化能.Liu等[19]通过磁控溅射制备了3种不同价态组合的Mg/(Nb2O5 + Nb)、Mg/Nb2O5以及Mg/Nb多层膜,发现Mg/(Nb2O5 + Nb)具有最好的吸/放氢性能,在225℃下仅需3 min就可吸氢4.9%,50 min可脱氢2.6%.并通过透射电镜(TEM)、X射线衍射仪(XRD)和X射线光电子能谱(XPS)得到了多价态铌离子作为Mg/Mg2+和H/H-之间电子转移的载体的直接证据. ...
... [18]所示.这是由于多相多价态的钛基催化剂可以作为Mg/MgH2吸/放氢过程中电子转移的载体,如图1b[18]所示,通过多价态的钛催化完成吸/放氢反应过程中的电子转移要比Mg和H或Mg2+和H-间直接转移电子更加容易,从而降低了反应活化能.Liu等[19]通过磁控溅射制备了3种不同价态组合的Mg/(Nb2O5 + Nb)、Mg/Nb2O5以及Mg/Nb多层膜,发现Mg/(Nb2O5 + Nb)具有最好的吸/放氢性能,在225℃下仅需3 min就可吸氢4.9%,50 min可脱氢2.6%.并通过透射电镜(TEM)、X射线衍射仪(XRD)和X射线光电子能谱(XPS)得到了多价态铌离子作为Mg/Mg2+和H/H-之间电子转移的载体的直接证据. ...
... [18]所示,通过多价态的钛催化完成吸/放氢反应过程中的电子转移要比Mg和H或Mg2+和H-间直接转移电子更加容易,从而降低了反应活化能.Liu等[19]通过磁控溅射制备了3种不同价态组合的Mg/(Nb2O5 + Nb)、Mg/Nb2O5以及Mg/Nb多层膜,发现Mg/(Nb2O5 + Nb)具有最好的吸/放氢性能,在225℃下仅需3 min就可吸氢4.9%,50 min可脱氢2.6%.并通过透射电镜(TEM)、X射线衍射仪(XRD)和X射线光电子能谱(XPS)得到了多价态铌离子作为Mg/Mg2+和H/H-之间电子转移的载体的直接证据. ...
... [
18]
Temperature programme desorption (TPD) profiles for the ball-milled (BM) MgH<sub>2</sub>, fully hydrogenated Mg-TiCl<sub>3</sub> and reaction-Mg-TiCl<sub>3</sub> sample (a), and the schematic diagram of the catalytic mechanism in de-/hydrogenation of BM-R (b) (<i>T</i>—temperature, <i>E</i><sub>A</sub>—activation energy, Δ<i>H</i>—enthalpy)<sup>[<xref ref-type="bibr" rid="R18">18</xref>]</sup>Fig.1![]()
除了多价态电子转移催化机制,过渡金属催化剂的电负性也是影响催化效果的重要因素.Cui等[20]将前述Mg-Ti核壳结构的复合材料扩展到Mg-TM (TM:Ti、Nb、V、Co、Mo或Ni)体系,发现随着TM自身电负性的减小,其提升MgH2脱氢动力学效果增强.这是因为当金属的电负性越大时,金属就越难失去电子,即TM—H键越弱;反之随着金属电负性的减小,金属越能束缚H.当金属电负性越接近于Mg—H电负性时,TM—H对MgH2中的Mg—H扰动也就越大,越能弱化Mg—H键的强度,导致反应动力学性能越快.但是对于金属Ni例外,这是因为其吸/放氢过程中形成Mg2NiH4/Mg2Ni,导致实际起催化作用的Ni显著减少. ...
... [
18]
Fig.1![]()
除了多价态电子转移催化机制,过渡金属催化剂的电负性也是影响催化效果的重要因素.Cui等[20]将前述Mg-Ti核壳结构的复合材料扩展到Mg-TM (TM:Ti、Nb、V、Co、Mo或Ni)体系,发现随着TM自身电负性的减小,其提升MgH2脱氢动力学效果增强.这是因为当金属的电负性越大时,金属就越难失去电子,即TM—H键越弱;反之随着金属电负性的减小,金属越能束缚H.当金属电负性越接近于Mg—H电负性时,TM—H对MgH2中的Mg—H扰动也就越大,越能弱化Mg—H键的强度,导致反应动力学性能越快.但是对于金属Ni例外,这是因为其吸/放氢过程中形成Mg2NiH4/Mg2Ni,导致实际起催化作用的Ni显著减少. ...
Direct microstructural evidence on the catalyzing mechanism for de/hydrogenation of Mg by multi-valence NbOx
1
2020
... 为进一步揭示多价态过渡金属及其化合物的催化机理,并获得更好的催化效果,Cui等[18]将球磨预处理后的Mg粉与过渡金属氯化物TiCl3在四氢呋喃(C4H8O)中反应制备了多相多价态钛基催化剂包覆的Mg颗粒,其脱氢活化能降至30.8 kJ/mol,起始脱氢温度约为175℃,比球磨MgH2、Mg-TiCl3材料分别降低了105和60℃,如图1a[18]所示.这是由于多相多价态的钛基催化剂可以作为Mg/MgH2吸/放氢过程中电子转移的载体,如图1b[18]所示,通过多价态的钛催化完成吸/放氢反应过程中的电子转移要比Mg和H或Mg2+和H-间直接转移电子更加容易,从而降低了反应活化能.Liu等[19]通过磁控溅射制备了3种不同价态组合的Mg/(Nb2O5 + Nb)、Mg/Nb2O5以及Mg/Nb多层膜,发现Mg/(Nb2O5 + Nb)具有最好的吸/放氢性能,在225℃下仅需3 min就可吸氢4.9%,50 min可脱氢2.6%.并通过透射电镜(TEM)、X射线衍射仪(XRD)和X射线光电子能谱(XPS)得到了多价态铌离子作为Mg/Mg2+和H/H-之间电子转移的载体的直接证据. ...
Mg-TM (TM: Ti, Nb, V, Co, Mo, or Ni) core-hell like nanostructures: Synthesis, hydrogen storage performance and catalytic mechanism
1
2014
... 除了多价态电子转移催化机制,过渡金属催化剂的电负性也是影响催化效果的重要因素.Cui等[20]将前述Mg-Ti核壳结构的复合材料扩展到Mg-TM (TM:Ti、Nb、V、Co、Mo或Ni)体系,发现随着TM自身电负性的减小,其提升MgH2脱氢动力学效果增强.这是因为当金属的电负性越大时,金属就越难失去电子,即TM—H键越弱;反之随着金属电负性的减小,金属越能束缚H.当金属电负性越接近于Mg—H电负性时,TM—H对MgH2中的Mg—H扰动也就越大,越能弱化Mg—H键的强度,导致反应动力学性能越快.但是对于金属Ni例外,这是因为其吸/放氢过程中形成Mg2NiH4/Mg2Ni,导致实际起催化作用的Ni显著减少. ...
In situ synthesized one-dimensional porous Ni@C nanorods as catalysts for hydrogen storage properties of MgH2
1
2014
... 此外,减小催化剂尺寸和提高其分散性可以进一步增强其催化作用[21].减小尺寸会增加催化活性位点,而增加催化剂分散度可以提高有效催化面积[22,23].例如,Chen等[22]利用单嘴静电纺丝技术制备了平均直径50 nm的多孔镍纳米纤维,将其添加到MgH2,得到的材料实现了143℃开始脱氢,325℃、11 min内释放7.02%的H2,脱氢活化能降低至81.5 kJ/mol.Wang等[24]将湿化学法刻蚀得到的NbTiC二维层状材料加入到MgH2中球磨,原位生成了超细的5 nm双金属NbTi纳米晶体,为MgH2的吸/放氢反应提供了大量催化活性位点.添加9%NbTiC的MgH2复合材料在195℃便开始脱氢,250℃下30 min内可释放5.8%的H2. ...
Porous Ni nanofibers with enhanced catalytic effect on the hydrogen storage performance of MgH2
2
2015
... 此外,减小催化剂尺寸和提高其分散性可以进一步增强其催化作用[21].减小尺寸会增加催化活性位点,而增加催化剂分散度可以提高有效催化面积[22,23].例如,Chen等[22]利用单嘴静电纺丝技术制备了平均直径50 nm的多孔镍纳米纤维,将其添加到MgH2,得到的材料实现了143℃开始脱氢,325℃、11 min内释放7.02%的H2,脱氢活化能降低至81.5 kJ/mol.Wang等[24]将湿化学法刻蚀得到的NbTiC二维层状材料加入到MgH2中球磨,原位生成了超细的5 nm双金属NbTi纳米晶体,为MgH2的吸/放氢反应提供了大量催化活性位点.添加9%NbTiC的MgH2复合材料在195℃便开始脱氢,250℃下30 min内可释放5.8%的H2. ...
... [22]利用单嘴静电纺丝技术制备了平均直径50 nm的多孔镍纳米纤维,将其添加到MgH2,得到的材料实现了143℃开始脱氢,325℃、11 min内释放7.02%的H2,脱氢活化能降低至81.5 kJ/mol.Wang等[24]将湿化学法刻蚀得到的NbTiC二维层状材料加入到MgH2中球磨,原位生成了超细的5 nm双金属NbTi纳米晶体,为MgH2的吸/放氢反应提供了大量催化活性位点.添加9%NbTiC的MgH2复合材料在195℃便开始脱氢,250℃下30 min内可释放5.8%的H2. ...
High-loading, ultrafine Ni nanoparticles dispersed on porous hollow carbon nanospheres for fast (de)hydrogenation kinetics of MgH2
1
2021
... 此外,减小催化剂尺寸和提高其分散性可以进一步增强其催化作用[21].减小尺寸会增加催化活性位点,而增加催化剂分散度可以提高有效催化面积[22,23].例如,Chen等[22]利用单嘴静电纺丝技术制备了平均直径50 nm的多孔镍纳米纤维,将其添加到MgH2,得到的材料实现了143℃开始脱氢,325℃、11 min内释放7.02%的H2,脱氢活化能降低至81.5 kJ/mol.Wang等[24]将湿化学法刻蚀得到的NbTiC二维层状材料加入到MgH2中球磨,原位生成了超细的5 nm双金属NbTi纳米晶体,为MgH2的吸/放氢反应提供了大量催化活性位点.添加9%NbTiC的MgH2复合材料在195℃便开始脱氢,250℃下30 min内可释放5.8%的H2. ...
In situ formed ultrafine NbTi nanocrystals from a NbTiC solid-solution MXene for hydrogen storage in MgH2
1
2019
... 此外,减小催化剂尺寸和提高其分散性可以进一步增强其催化作用[21].减小尺寸会增加催化活性位点,而增加催化剂分散度可以提高有效催化面积[22,23].例如,Chen等[22]利用单嘴静电纺丝技术制备了平均直径50 nm的多孔镍纳米纤维,将其添加到MgH2,得到的材料实现了143℃开始脱氢,325℃、11 min内释放7.02%的H2,脱氢活化能降低至81.5 kJ/mol.Wang等[24]将湿化学法刻蚀得到的NbTiC二维层状材料加入到MgH2中球磨,原位生成了超细的5 nm双金属NbTi纳米晶体,为MgH2的吸/放氢反应提供了大量催化活性位点.添加9%NbTiC的MgH2复合材料在195℃便开始脱氢,250℃下30 min内可释放5.8%的H2. ...
Watching in situ the hydrogen diffusion dynamics in magnesium on the nanoscale
1
2020
... 纳米化/纳米限域也是改善MgH2吸/放氢性能的有效手段.Karst等[25]通过对Mg自支撑薄膜的原位光学成像观察定性地揭示了金属Mg向MgH2转变的相变动力学过程,MgH2在Mg晶界处成核,且在随后的氢化过程中先形成的氢化物是后续反应形核生长的成核中心,氢化过程是从一个晶粒扩展另一个晶粒,直到在整个薄膜表面形成MgH2,即在每一个Mg晶粒转变为MgH2之前都需要新的成核点.随着氢化的进行,Mg薄膜内部形成的MgH2层减缓了薄膜氢化过程,吸氢显著变慢.有趣的是,薄膜中单个的晶粒在短短几分钟内就被完全氢化,表明调整晶粒尺寸和增加晶界或成核位点的数量是加快氢化的有效途径,对储氢性能有重要影响.Cui等[26]通过模型构建,从理论上也说明了随着材料颗粒/粒度的变化,脱氢反应的熵(ΔS)也会随之发生改变,进而影响MgH2的脱氢温度.Ouyang等[27]以纳米Mg2.9Ni薄膜为研究对象,引入界面能项对van't Hoff方程进行修正,并采用Miedema半经验方程估算界面能.由修正方程可知,当晶粒或颗粒尺寸小于2 nm时,MgH2的脱氢热力学将发生显著改变,同时从实验上验证了该修正结果的有效性.目前普遍认为,与微米尺度的MgH2相比,纳米化的MgH2拥有更大的比表面积和更多的晶界,从而为Mg/MgH2吸/放氢反应提供了更多的氢扩散通道和反应活性位点.而纳米化的MgH2自身所储存的额外的界面能,也会降低MgH2的热力学稳定性.因此,纳米化的MgH2可以实现热力学和动力学双调控,显著改善储氢性能. ...
On the hydrogen desorption entropy change of modified MgH2
1
2018
... 纳米化/纳米限域也是改善MgH2吸/放氢性能的有效手段.Karst等[25]通过对Mg自支撑薄膜的原位光学成像观察定性地揭示了金属Mg向MgH2转变的相变动力学过程,MgH2在Mg晶界处成核,且在随后的氢化过程中先形成的氢化物是后续反应形核生长的成核中心,氢化过程是从一个晶粒扩展另一个晶粒,直到在整个薄膜表面形成MgH2,即在每一个Mg晶粒转变为MgH2之前都需要新的成核点.随着氢化的进行,Mg薄膜内部形成的MgH2层减缓了薄膜氢化过程,吸氢显著变慢.有趣的是,薄膜中单个的晶粒在短短几分钟内就被完全氢化,表明调整晶粒尺寸和增加晶界或成核位点的数量是加快氢化的有效途径,对储氢性能有重要影响.Cui等[26]通过模型构建,从理论上也说明了随着材料颗粒/粒度的变化,脱氢反应的熵(ΔS)也会随之发生改变,进而影响MgH2的脱氢温度.Ouyang等[27]以纳米Mg2.9Ni薄膜为研究对象,引入界面能项对van't Hoff方程进行修正,并采用Miedema半经验方程估算界面能.由修正方程可知,当晶粒或颗粒尺寸小于2 nm时,MgH2的脱氢热力学将发生显著改变,同时从实验上验证了该修正结果的有效性.目前普遍认为,与微米尺度的MgH2相比,纳米化的MgH2拥有更大的比表面积和更多的晶界,从而为Mg/MgH2吸/放氢反应提供了更多的氢扩散通道和反应活性位点.而纳米化的MgH2自身所储存的额外的界面能,也会降低MgH2的热力学稳定性.因此,纳米化的MgH2可以实现热力学和动力学双调控,显著改善储氢性能. ...
Effect of interfacial free energy on hydriding reaction of Mg-Ni thin films
1
2007
... 纳米化/纳米限域也是改善MgH2吸/放氢性能的有效手段.Karst等[25]通过对Mg自支撑薄膜的原位光学成像观察定性地揭示了金属Mg向MgH2转变的相变动力学过程,MgH2在Mg晶界处成核,且在随后的氢化过程中先形成的氢化物是后续反应形核生长的成核中心,氢化过程是从一个晶粒扩展另一个晶粒,直到在整个薄膜表面形成MgH2,即在每一个Mg晶粒转变为MgH2之前都需要新的成核点.随着氢化的进行,Mg薄膜内部形成的MgH2层减缓了薄膜氢化过程,吸氢显著变慢.有趣的是,薄膜中单个的晶粒在短短几分钟内就被完全氢化,表明调整晶粒尺寸和增加晶界或成核位点的数量是加快氢化的有效途径,对储氢性能有重要影响.Cui等[26]通过模型构建,从理论上也说明了随着材料颗粒/粒度的变化,脱氢反应的熵(ΔS)也会随之发生改变,进而影响MgH2的脱氢温度.Ouyang等[27]以纳米Mg2.9Ni薄膜为研究对象,引入界面能项对van't Hoff方程进行修正,并采用Miedema半经验方程估算界面能.由修正方程可知,当晶粒或颗粒尺寸小于2 nm时,MgH2的脱氢热力学将发生显著改变,同时从实验上验证了该修正结果的有效性.目前普遍认为,与微米尺度的MgH2相比,纳米化的MgH2拥有更大的比表面积和更多的晶界,从而为Mg/MgH2吸/放氢反应提供了更多的氢扩散通道和反应活性位点.而纳米化的MgH2自身所储存的额外的界面能,也会降低MgH2的热力学稳定性.因此,纳米化的MgH2可以实现热力学和动力学双调控,显著改善储氢性能. ...
Monodisperse magnesium hydride nanoparticles uniformly self-assembled on graphene
1
2015
... 纳米化可以是通过直接制备使储氢材料自身纳米化(无负载型),也可以是将之装载到载体的纳米空隙内(纳米限域、负载型).这2种纳米化方法都得到了广泛重视.纳米限域不仅可以获得分布均匀、尺寸较小的纳米颗粒Mg/MgH2,还可以有效抑制其在吸/放氢反应过程中的团聚长大.Xia等[28]通过高压溶剂热反应实现了MgH2在石墨烯表面的高负载自组装可控合成,MgH2的负载量由通常的20%~30%提高到75%,同时,以金属镍烷基有机物为前驱体实现了Ni的原位掺杂,Ni纳米颗粒修饰的石墨烯表面负载量达75%的MgH2 (Ni-MHGH-75)纳米复合材料脱氢温度可降低至201℃,储氢容量高达5.4%,在200℃时Ni-MHGH-75在10 min内完全氢化,并且在100 cyc循环过程中,容量保持率达99.2%.随后Shinde等[29]采用类似的方法,用水合肼替代H2作为还原剂,制备了三维碳材料纳米限域和过渡金属修饰的MgH2纳米复合材料,可逆储氢容量达6.63%,在180℃时5 min即可完成吸氢,20 min内放氢6.6%.该材料在空气中具有一定的稳定性,且在180℃长达435 h的100 cyc循环中,可逆储氢容量和反应动力学性能均没有明显衰减,容量保持率高达99.56%. ...
Self-assembled air-stable magnesium hydride embedded in 3-D activated carbon for reversible hydrogen storage
1
2017
... 纳米化可以是通过直接制备使储氢材料自身纳米化(无负载型),也可以是将之装载到载体的纳米空隙内(纳米限域、负载型).这2种纳米化方法都得到了广泛重视.纳米限域不仅可以获得分布均匀、尺寸较小的纳米颗粒Mg/MgH2,还可以有效抑制其在吸/放氢反应过程中的团聚长大.Xia等[28]通过高压溶剂热反应实现了MgH2在石墨烯表面的高负载自组装可控合成,MgH2的负载量由通常的20%~30%提高到75%,同时,以金属镍烷基有机物为前驱体实现了Ni的原位掺杂,Ni纳米颗粒修饰的石墨烯表面负载量达75%的MgH2 (Ni-MHGH-75)纳米复合材料脱氢温度可降低至201℃,储氢容量高达5.4%,在200℃时Ni-MHGH-75在10 min内完全氢化,并且在100 cyc循环过程中,容量保持率达99.2%.随后Shinde等[29]采用类似的方法,用水合肼替代H2作为还原剂,制备了三维碳材料纳米限域和过渡金属修饰的MgH2纳米复合材料,可逆储氢容量达6.63%,在180℃时5 min即可完成吸氢,20 min内放氢6.6%.该材料在空气中具有一定的稳定性,且在180℃长达435 h的100 cyc循环中,可逆储氢容量和反应动力学性能均没有明显衰减,容量保持率高达99.56%. ...
Synthesis of metallic magnesium nanoparticles by sonoelectrochemistry
1
2008
... 直接合成纳米Mg/MgH2往往需要比较复杂的制备技术,并且纳米Mg/MgH2往往会在较高温度下的吸/放氢循环过程中迅速长大[30].最近,Zhang等[31]将球磨预处理后的MgCl2与LiH在四氢呋喃中搅拌反应,然后对得到的有机悬浊液进行超声处理,接着对产物进行过滤、清洗、离心和干燥后,得到了尺寸为4~5 nm的超细MgH2颗粒.得益于自身超细小的粒度以及未使用多余的限域模板,其热力学稳定性得以降低且动力学提高,获得的纳米颗粒在30℃时可逆储氢容量为6.7%,并且在150℃条件下的50 cyc循环中保持了稳定且快速的循环吸/放氢性能.该方法相对简单,超声波在液体介质中产生的空化效应所提供的能量促进了MgCl2与LiH之间的接触和反应,同时超声波的破碎效果也能够有效抑制MgH2的长大,避免了冗余载体的添加. ...
Realizing 6.7 wt% reversible storage of hydrogen at ambient temperature with non-confined ultrafine magnesium hydrides
1
2021
... 直接合成纳米Mg/MgH2往往需要比较复杂的制备技术,并且纳米Mg/MgH2往往会在较高温度下的吸/放氢循环过程中迅速长大[30].最近,Zhang等[31]将球磨预处理后的MgCl2与LiH在四氢呋喃中搅拌反应,然后对得到的有机悬浊液进行超声处理,接着对产物进行过滤、清洗、离心和干燥后,得到了尺寸为4~5 nm的超细MgH2颗粒.得益于自身超细小的粒度以及未使用多余的限域模板,其热力学稳定性得以降低且动力学提高,获得的纳米颗粒在30℃时可逆储氢容量为6.7%,并且在150℃条件下的50 cyc循环中保持了稳定且快速的循环吸/放氢性能.该方法相对简单,超声波在液体介质中产生的空化效应所提供的能量促进了MgCl2与LiH之间的接触和反应,同时超声波的破碎效果也能够有效抑制MgH2的长大,避免了冗余载体的添加. ...
Microstructure and hydrogen absorption properties of nano-phase composite prepared by mechanical alloying of MmNi5-x(CoAlMn)x and Mg
1
1999
... 自催化作用是一种利用动力学性能好的储氢合金(如AB5、TiFe合金等)来改善动力学性能较差的MgH2的吸/放氢动力学的机制.Zhu等[32~37]用高能球磨法和薄膜技术制备了多种复合储氢合金.以Mg/MmNi5-x(CoAlMn)x (简记为MmNi5,Mm代表混合稀土)纳米晶复合储氢合金为例,球磨使Mg与MmNi5化合形成La2Mg17以及Mg2Ni等活性相.Mg的含量对于复合储氢合金的晶粒尺寸有显著影响,即随着Mg含量的增加,相同球磨条件下制备的合金中MmNi5的晶粒尺寸增加.该方法制备的纳米晶复合储氢合金中拥有极高密度的界面(包括晶界与相界),其活化以及吸氢性能较铸态合金有比较大的提高,无需活化或只需活化1次即可吸氢.对吸氢动力学曲线拟合表明,Mg/MmNi5纳米复合合金的吸氢符合自催化机制[38],MmNi5的催化作用和高密度相界提供的氢扩散通道提高了合金的吸氢动力学.同样地,将氢化、快淬处理以及未处理的原始铸锭Ti0.4Cr0.15Mn0.15V0.3合金(以下统称BCC合金)与MgH2球磨得到复合材料,所得复合材料的储氢性能催化效果为氢化BCC合金>快淬BCC合金>铸锭BCC合金.这主要是因为BCC合金氢化后体积膨胀导致形成裂纹或者破碎,这些粒径较小的微/纳米级别的氢化BCC合金与MgH2复合后,提供更多氢扩散通道的同时还能加速氢分子的解离和重组[39]. ...
Composite structure and hydrogen storage properties in Mg-base alloys
0
2006
Microstructure of MmM5/Mg multi-layer films prepared by magnetron sputtering
0
2005
MmM5/Mg multi-layer hydrogen storage thin films prepared by dc magnetron sputtering
0
2004
Microstructure and hydrogen sorption properties of Mg–Ni/MmM5 multi-layer film by magne-tron sputtering
0
2004
Microstructure of MmM5/Mg multi-layer hydrogen storage films prepared by magnetron sputtering
1
2004
... 自催化作用是一种利用动力学性能好的储氢合金(如AB5、TiFe合金等)来改善动力学性能较差的MgH2的吸/放氢动力学的机制.Zhu等[32~37]用高能球磨法和薄膜技术制备了多种复合储氢合金.以Mg/MmNi5-x(CoAlMn)x (简记为MmNi5,Mm代表混合稀土)纳米晶复合储氢合金为例,球磨使Mg与MmNi5化合形成La2Mg17以及Mg2Ni等活性相.Mg的含量对于复合储氢合金的晶粒尺寸有显著影响,即随着Mg含量的增加,相同球磨条件下制备的合金中MmNi5的晶粒尺寸增加.该方法制备的纳米晶复合储氢合金中拥有极高密度的界面(包括晶界与相界),其活化以及吸氢性能较铸态合金有比较大的提高,无需活化或只需活化1次即可吸氢.对吸氢动力学曲线拟合表明,Mg/MmNi5纳米复合合金的吸氢符合自催化机制[38],MmNi5的催化作用和高密度相界提供的氢扩散通道提高了合金的吸氢动力学.同样地,将氢化、快淬处理以及未处理的原始铸锭Ti0.4Cr0.15Mn0.15V0.3合金(以下统称BCC合金)与MgH2球磨得到复合材料,所得复合材料的储氢性能催化效果为氢化BCC合金>快淬BCC合金>铸锭BCC合金.这主要是因为BCC合金氢化后体积膨胀导致形成裂纹或者破碎,这些粒径较小的微/纳米级别的氢化BCC合金与MgH2复合后,提供更多氢扩散通道的同时还能加速氢分子的解离和重组[39]. ...
Hydriding kinetics of nano-phase composite hydrogen storage alloys prepared by mechanical alloying of Mg and MmNi5-x(CoAlMn)x
1
2002
... 自催化作用是一种利用动力学性能好的储氢合金(如AB5、TiFe合金等)来改善动力学性能较差的MgH2的吸/放氢动力学的机制.Zhu等[32~37]用高能球磨法和薄膜技术制备了多种复合储氢合金.以Mg/MmNi5-x(CoAlMn)x (简记为MmNi5,Mm代表混合稀土)纳米晶复合储氢合金为例,球磨使Mg与MmNi5化合形成La2Mg17以及Mg2Ni等活性相.Mg的含量对于复合储氢合金的晶粒尺寸有显著影响,即随着Mg含量的增加,相同球磨条件下制备的合金中MmNi5的晶粒尺寸增加.该方法制备的纳米晶复合储氢合金中拥有极高密度的界面(包括晶界与相界),其活化以及吸氢性能较铸态合金有比较大的提高,无需活化或只需活化1次即可吸氢.对吸氢动力学曲线拟合表明,Mg/MmNi5纳米复合合金的吸氢符合自催化机制[38],MmNi5的催化作用和高密度相界提供的氢扩散通道提高了合金的吸氢动力学.同样地,将氢化、快淬处理以及未处理的原始铸锭Ti0.4Cr0.15Mn0.15V0.3合金(以下统称BCC合金)与MgH2球磨得到复合材料,所得复合材料的储氢性能催化效果为氢化BCC合金>快淬BCC合金>铸锭BCC合金.这主要是因为BCC合金氢化后体积膨胀导致形成裂纹或者破碎,这些粒径较小的微/纳米级别的氢化BCC合金与MgH2复合后,提供更多氢扩散通道的同时还能加速氢分子的解离和重组[39]. ...
The effect of a Ti-V-based BCC alloy as a catalyst on the hydrogen storage properties of MgH2
1
2010
... 自催化作用是一种利用动力学性能好的储氢合金(如AB5、TiFe合金等)来改善动力学性能较差的MgH2的吸/放氢动力学的机制.Zhu等[32~37]用高能球磨法和薄膜技术制备了多种复合储氢合金.以Mg/MmNi5-x(CoAlMn)x (简记为MmNi5,Mm代表混合稀土)纳米晶复合储氢合金为例,球磨使Mg与MmNi5化合形成La2Mg17以及Mg2Ni等活性相.Mg的含量对于复合储氢合金的晶粒尺寸有显著影响,即随着Mg含量的增加,相同球磨条件下制备的合金中MmNi5的晶粒尺寸增加.该方法制备的纳米晶复合储氢合金中拥有极高密度的界面(包括晶界与相界),其活化以及吸氢性能较铸态合金有比较大的提高,无需活化或只需活化1次即可吸氢.对吸氢动力学曲线拟合表明,Mg/MmNi5纳米复合合金的吸氢符合自催化机制[38],MmNi5的催化作用和高密度相界提供的氢扩散通道提高了合金的吸氢动力学.同样地,将氢化、快淬处理以及未处理的原始铸锭Ti0.4Cr0.15Mn0.15V0.3合金(以下统称BCC合金)与MgH2球磨得到复合材料,所得复合材料的储氢性能催化效果为氢化BCC合金>快淬BCC合金>铸锭BCC合金.这主要是因为BCC合金氢化后体积膨胀导致形成裂纹或者破碎,这些粒径较小的微/纳米级别的氢化BCC合金与MgH2复合后,提供更多氢扩散通道的同时还能加速氢分子的解离和重组[39]. ...
Cooperative hydriding properties in a nanostructured Mg2Ni-H system
1
1997
... 多相复合体系中组元之间在吸/放氢反应过程中应变作用诱发协同放氢亦是一种重要的机制.Fujii等[40]研究了1 MPa氢压球磨Mg2Ni合金的储氢性能,球磨60 min的合金可以在453 K、30 min释放出理论容量中90%的H2,而材料能够低温放氢的关键是不同晶粒之间的晶格收缩产生的应力.随后,Zaluska等[41]制备了MgH2-35%Mg2NiH4复合材料,可于220~240℃脱氢.他们也认为,当Mg2NiH4在低温下放氢时晶格收缩产生的应力作用于相邻的MgH2,而后脱氢的MgH2同样发生了晶格收缩,产生的应力再作用于其相邻的Mg2NiH4,多相复合结构在吸/放氢反应过程中的应力协同效应导致了吸/放氢温度的降低,提高了脱氢动力学性能.Higuchi等[42]用射频磁控溅射制备的Pd/Mg/Pd三层膜可以在100℃条件下实现可逆放氢,并认为主要是吸氢后的Pd/Mg/Pd三层膜在Pd膜开始脱氢后晶格收缩产生的应力(收缩力)作用于处于中间夹层的Mg膜,从而使Mg膜中的氢更容易脱出,即应力协同是实现低温可逆吸/放氢的原因. ...
Synergy of hydrogen sorption in ball-milled hydrides of Mg and Mg2Ni
1
1999
... 多相复合体系中组元之间在吸/放氢反应过程中应变作用诱发协同放氢亦是一种重要的机制.Fujii等[40]研究了1 MPa氢压球磨Mg2Ni合金的储氢性能,球磨60 min的合金可以在453 K、30 min释放出理论容量中90%的H2,而材料能够低温放氢的关键是不同晶粒之间的晶格收缩产生的应力.随后,Zaluska等[41]制备了MgH2-35%Mg2NiH4复合材料,可于220~240℃脱氢.他们也认为,当Mg2NiH4在低温下放氢时晶格收缩产生的应力作用于相邻的MgH2,而后脱氢的MgH2同样发生了晶格收缩,产生的应力再作用于其相邻的Mg2NiH4,多相复合结构在吸/放氢反应过程中的应力协同效应导致了吸/放氢温度的降低,提高了脱氢动力学性能.Higuchi等[42]用射频磁控溅射制备的Pd/Mg/Pd三层膜可以在100℃条件下实现可逆放氢,并认为主要是吸氢后的Pd/Mg/Pd三层膜在Pd膜开始脱氢后晶格收缩产生的应力(收缩力)作用于处于中间夹层的Mg膜,从而使Mg膜中的氢更容易脱出,即应力协同是实现低温可逆吸/放氢的原因. ...
Remarkable hydrogen storage properties in three-layered Pd/Mg/Pd thin films
1
2002
... 多相复合体系中组元之间在吸/放氢反应过程中应变作用诱发协同放氢亦是一种重要的机制.Fujii等[40]研究了1 MPa氢压球磨Mg2Ni合金的储氢性能,球磨60 min的合金可以在453 K、30 min释放出理论容量中90%的H2,而材料能够低温放氢的关键是不同晶粒之间的晶格收缩产生的应力.随后,Zaluska等[41]制备了MgH2-35%Mg2NiH4复合材料,可于220~240℃脱氢.他们也认为,当Mg2NiH4在低温下放氢时晶格收缩产生的应力作用于相邻的MgH2,而后脱氢的MgH2同样发生了晶格收缩,产生的应力再作用于其相邻的Mg2NiH4,多相复合结构在吸/放氢反应过程中的应力协同效应导致了吸/放氢温度的降低,提高了脱氢动力学性能.Higuchi等[42]用射频磁控溅射制备的Pd/Mg/Pd三层膜可以在100℃条件下实现可逆放氢,并认为主要是吸氢后的Pd/Mg/Pd三层膜在Pd膜开始脱氢后晶格收缩产生的应力(收缩力)作用于处于中间夹层的Mg膜,从而使Mg膜中的氢更容易脱出,即应力协同是实现低温可逆吸/放氢的原因. ...
Enhanced hydrogen storage kinetics and stability by synergistic effects of in situ formed CeH2.73 and Ni in CeH2.73-MgH2-Ni nanocomposites
5
2014
... 然而,上述方法难以满足储氢合金规模应用的要求.发展实现上述机制的合金的规模化制备方法十分关键.我们近年来发展一种通过氢化反应原位生成纳米多相复合结构合金的规模制备方法.例如:采用熔炼方法得到Mg3Ce化合物,并通过氢致歧化反应原位生成了MgH2-CeH2.73纳米复合材料[43].在合金中进一步加入金属Ni,获得了MgH2和CeH2.73呈层状排列、Ni分布于两相界面的独特结构,如图2a和b[43]给出的明场像和电子衍射图所示.CeH2.73、Ni催化相同时存在大大提高了多相复合后的界面面积,其所产生的协同效应和大量的界面能提高了材料的吸/放氢动力学性能,Mg80Ce18Ni2合金在232℃脱氢,可逆容量> 4.0%,500 cyc循环后容量保持率高于80%,如图2c[43]所示. ...
... [43]给出的明场像和电子衍射图所示.CeH2.73、Ni催化相同时存在大大提高了多相复合后的界面面积,其所产生的协同效应和大量的界面能提高了材料的吸/放氢动力学性能,Mg80Ce18Ni2合金在232℃脱氢,可逆容量> 4.0%,500 cyc循环后容量保持率高于80%,如图2c[43]所示. ...
... [43]所示. ...
... [
43]
(a) bright field image of the in situ formed CeH2.73-MgH2-Ni composite ...
... (c) evolution of the maximum hydrogen sorption capacities versus cycle times of CeH
2.73-MgH
2-Ni composite
TEM image and analyses of the microstructure of the partially dehydrogenated CeH<sub>2.73</sub>-MgH<sub>2</sub>-Ni nanocomposites demonstrating the catalyst effect of CeH<sub>2.73</sub> and Ni on MgH<sub>2</sub> dehydrogenation process<sup>[<xref ref-type="bibr" rid="R43">43</xref>]</sup>Fig.2![]()
将快淬法与氢化原位生成纳米相的方法结合也是一种高效规模制备纳米复合储氢合金的方法.Luo等[44]使用快淬法制备了Mg80Nd4Ni8合金,氢化后原位生成NdH2-Mg-Mg2Ni纳米复合材料,该材料在300℃经过819 cyc循环后保持3.75%的可逆容量,容量保持率为78.6%.这主要归因于原位形成的Mg-NdH2-Mg2Ni纳米复合材料中存在的大量晶界为H原子扩散提供了通道且生成的纳米NdH2提升了反应动力学.此外,Mg2Ni和NdH2也起到了抑制Mg基体晶粒长大的作用,维持了材料初始状态的相结构和纳米尺度.Lin等[45]通过快淬的方法制备了Mg-Ce-Ni非晶合金,该非晶合金氢化后原位形成了MgH2-Mg2NiH4-CeH2.73多相复合结构.该复合材料在室温下3 min吸氢2.7%,最大储氢容量为3.1%,最低脱氢温度为224℃.原位生成的Mg2Ni以及稳定存在的LaH2提供了足够的相界和晶界来加快氢的扩散速率,同时,CeH3还具有自催化效应和协同效应,因此提高了MgH2的吸/放氢动力学.进一步研究发现,对纳米MgH2-Mg2NiH4-CeH2.73复合物进行控制氧化处理,CeH2.73原位转变为核壳结构的CeH2.73/CeO2,如图3a[46]所示,进一步显著降低了放氢温度.图3b[46]所示为TEM观察到的核壳结构的纳米CeH2.73/CeO2共生相,原位HRTEM观察和第一性原理计算证明CeH2.73/CeO2界面处的低能垒氢逸出效应是实现脱氢动力学提高的主要原因,即纳米CeH2.73/CeO2共生相起到类似“氢泵”的作用[46],显著降低了MgH2的脱氢温度. ...
Achieving superior cycling stability by in situ forming NdH2-Mg-Mg2Ni nanocomposites
1
2018
... 将快淬法与氢化原位生成纳米相的方法结合也是一种高效规模制备纳米复合储氢合金的方法.Luo等[44]使用快淬法制备了Mg80Nd4Ni8合金,氢化后原位生成NdH2-Mg-Mg2Ni纳米复合材料,该材料在300℃经过819 cyc循环后保持3.75%的可逆容量,容量保持率为78.6%.这主要归因于原位形成的Mg-NdH2-Mg2Ni纳米复合材料中存在的大量晶界为H原子扩散提供了通道且生成的纳米NdH2提升了反应动力学.此外,Mg2Ni和NdH2也起到了抑制Mg基体晶粒长大的作用,维持了材料初始状态的相结构和纳米尺度.Lin等[45]通过快淬的方法制备了Mg-Ce-Ni非晶合金,该非晶合金氢化后原位形成了MgH2-Mg2NiH4-CeH2.73多相复合结构.该复合材料在室温下3 min吸氢2.7%,最大储氢容量为3.1%,最低脱氢温度为224℃.原位生成的Mg2Ni以及稳定存在的LaH2提供了足够的相界和晶界来加快氢的扩散速率,同时,CeH3还具有自催化效应和协同效应,因此提高了MgH2的吸/放氢动力学.进一步研究发现,对纳米MgH2-Mg2NiH4-CeH2.73复合物进行控制氧化处理,CeH2.73原位转变为核壳结构的CeH2.73/CeO2,如图3a[46]所示,进一步显著降低了放氢温度.图3b[46]所示为TEM观察到的核壳结构的纳米CeH2.73/CeO2共生相,原位HRTEM观察和第一性原理计算证明CeH2.73/CeO2界面处的低能垒氢逸出效应是实现脱氢动力学提高的主要原因,即纳米CeH2.73/CeO2共生相起到类似“氢泵”的作用[46],显著降低了MgH2的脱氢温度. ...
Phase transition and hydrogen storage properties of melt-spun Mg3LaNi0.1 alloy
1
2012
... 将快淬法与氢化原位生成纳米相的方法结合也是一种高效规模制备纳米复合储氢合金的方法.Luo等[44]使用快淬法制备了Mg80Nd4Ni8合金,氢化后原位生成NdH2-Mg-Mg2Ni纳米复合材料,该材料在300℃经过819 cyc循环后保持3.75%的可逆容量,容量保持率为78.6%.这主要归因于原位形成的Mg-NdH2-Mg2Ni纳米复合材料中存在的大量晶界为H原子扩散提供了通道且生成的纳米NdH2提升了反应动力学.此外,Mg2Ni和NdH2也起到了抑制Mg基体晶粒长大的作用,维持了材料初始状态的相结构和纳米尺度.Lin等[45]通过快淬的方法制备了Mg-Ce-Ni非晶合金,该非晶合金氢化后原位形成了MgH2-Mg2NiH4-CeH2.73多相复合结构.该复合材料在室温下3 min吸氢2.7%,最大储氢容量为3.1%,最低脱氢温度为224℃.原位生成的Mg2Ni以及稳定存在的LaH2提供了足够的相界和晶界来加快氢的扩散速率,同时,CeH3还具有自催化效应和协同效应,因此提高了MgH2的吸/放氢动力学.进一步研究发现,对纳米MgH2-Mg2NiH4-CeH2.73复合物进行控制氧化处理,CeH2.73原位转变为核壳结构的CeH2.73/CeO2,如图3a[46]所示,进一步显著降低了放氢温度.图3b[46]所示为TEM观察到的核壳结构的纳米CeH2.73/CeO2共生相,原位HRTEM观察和第一性原理计算证明CeH2.73/CeO2界面处的低能垒氢逸出效应是实现脱氢动力学提高的主要原因,即纳米CeH2.73/CeO2共生相起到类似“氢泵”的作用[46],显著降低了MgH2的脱氢温度. ...
Symbiotic CeH2.73/CeO2 catalyst: A novel hydrogen pump
5
2014
... 将快淬法与氢化原位生成纳米相的方法结合也是一种高效规模制备纳米复合储氢合金的方法.Luo等[44]使用快淬法制备了Mg80Nd4Ni8合金,氢化后原位生成NdH2-Mg-Mg2Ni纳米复合材料,该材料在300℃经过819 cyc循环后保持3.75%的可逆容量,容量保持率为78.6%.这主要归因于原位形成的Mg-NdH2-Mg2Ni纳米复合材料中存在的大量晶界为H原子扩散提供了通道且生成的纳米NdH2提升了反应动力学.此外,Mg2Ni和NdH2也起到了抑制Mg基体晶粒长大的作用,维持了材料初始状态的相结构和纳米尺度.Lin等[45]通过快淬的方法制备了Mg-Ce-Ni非晶合金,该非晶合金氢化后原位形成了MgH2-Mg2NiH4-CeH2.73多相复合结构.该复合材料在室温下3 min吸氢2.7%,最大储氢容量为3.1%,最低脱氢温度为224℃.原位生成的Mg2Ni以及稳定存在的LaH2提供了足够的相界和晶界来加快氢的扩散速率,同时,CeH3还具有自催化效应和协同效应,因此提高了MgH2的吸/放氢动力学.进一步研究发现,对纳米MgH2-Mg2NiH4-CeH2.73复合物进行控制氧化处理,CeH2.73原位转变为核壳结构的CeH2.73/CeO2,如图3a[46]所示,进一步显著降低了放氢温度.图3b[46]所示为TEM观察到的核壳结构的纳米CeH2.73/CeO2共生相,原位HRTEM观察和第一性原理计算证明CeH2.73/CeO2界面处的低能垒氢逸出效应是实现脱氢动力学提高的主要原因,即纳米CeH2.73/CeO2共生相起到类似“氢泵”的作用[46],显著降低了MgH2的脱氢温度. ...
... [46]所示为TEM观察到的核壳结构的纳米CeH2.73/CeO2共生相,原位HRTEM观察和第一性原理计算证明CeH2.73/CeO2界面处的低能垒氢逸出效应是实现脱氢动力学提高的主要原因,即纳米CeH2.73/CeO2共生相起到类似“氢泵”的作用[46],显著降低了MgH2的脱氢温度. ...
... [46],显著降低了MgH2的脱氢温度. ...
... [
46]
(a) HRTEM image of typical symbiotic CeH2.73/CeO2 nanoparticles ...
... (c-e) HRTEM image showing the magnified area in Fig.3b (c); the insets are the corresponding fast Fourier transformation (FFT) patterns of the outer (d) and inner (e) layers of the core-shell structure (Zoon axis [011])
Microstructure characterizations<sup>[<xref ref-type="bibr" rid="R46">46</xref>]</sup>Fig.3![]()
MgH2与配位氢化物复合也是近年来研究的热点[47~50].这种多相复合体系虽然具有比较高的储氢容量,但由于添加了配位氢化物,形成了新的吸/放氢反应途径,降低了反应焓变同时也带来了动力学缓慢、特别是反应不可逆等问题,在此不再赘述. ...
Cooperative catalysis on the dehydrogenation of NdCl3 doped LiBH4-MgH2 composites
1
2011
... MgH2与配位氢化物复合也是近年来研究的热点[47~50].这种多相复合体系虽然具有比较高的储氢容量,但由于添加了配位氢化物,形成了新的吸/放氢反应途径,降低了反应焓变同时也带来了动力学缓慢、特别是反应不可逆等问题,在此不再赘述. ...
Enhanced dehydrogenation properties of LiBH4 compositing with hydrogenated magnesium-rare earth compounds
0
2012
Synergetic effects of hydrogenated Mg3La and TiCl3 on the dehydrogenation of LiBH4
0
2011
Direct synthesis and hydrogen storage characteristics of Mg-B-H compounds
2
2012
... MgH2与配位氢化物复合也是近年来研究的热点[47~50].这种多相复合体系虽然具有比较高的储氢容量,但由于添加了配位氢化物,形成了新的吸/放氢反应途径,降低了反应焓变同时也带来了动力学缓慢、特别是反应不可逆等问题,在此不再赘述. ...
... AB3型的LaMg2Ni9合金结构之一为如图4[52]所示的PuNi3型结构[53],其可看成是AB5结构单元(CaCu5型)和AB2结构单元(MgZn2型或MgCu2型)沿c轴堆叠而成.A2B7型RE-Mg-Ni系合金是在AB3型合金基础上发展起来的,只需改变沿c轴堆叠AB2单元和AB5结构单元比例即可,即A2B4 + 2 × AB5 = 2 × A2B7.由于Mg在RE-Mg-Ni基合金中固溶度有限,以及在合金中具有选择性占位,对合金的相组成有比较大的影响.一般来说,Mg原子只进入[A2B4]结构单元而不占据[AB5]单元.AB3型和A2B7型合金都具有比AB5合金更高的理论储氢量和对应的电化学容量[50,54].Kohno等[55]研究了La-Mg-Nix(x = 3~3.5)系列合金的电化学性能,发现x = 3.0、3.3和3.5的3种合金电极对应的放电容量分别为387、410和390 mAh/g,均高于LaNi5合金电极 320 mAh/g的容量.RE-Mg-Ni系合金高容量的优点,使之成为新一代Ni-MH电池负极材料.Pan等[56]研究了不同化学计量比La0.7Mg0.3(Ni0.85Co0.15)x(x = 2.5、3.0、3.5、4.0、5.0)合金的电化学性能,发现放电容量、高倍率放电性能、交换电流密度等均随x增加表现出先升后降的趋势.与之相对应的是,AB3主相的含量随x的增加而减少,AB5相含量恰恰相反.当x = 3.5时,合金由AB3主相和少量AB5相构成,这种多相结构合金电极的电化学性能最佳.该结果为后续开展多相复合以及多元合金化提供了重要启示. ...
The development of hydrogen storage alloys and the progress of nickel hydride batteries
1
2001
... Ni-MH电池负极材料是储氢合金在电化学储能领域的主要应用场景.为获得优良的工作表现,储氢合金负极材料应满足以下要求[51]:① 高的可逆储氢容量以及合适的吸/放氢平台压;② 对电化学吸/放氢具有很好的电催化作用; ③在碱性电解液中具有良好的耐腐蚀性能;④ 具有较宽的工作温度;⑤ 具有良好的动力学性能和循环性能;⑥ 资源丰富,价格低廉,无污染.过去普遍使用的AB5合金存在容量低的短板,近年来主要发展高容量的A2B7型或AB3型稀土(RE)-Mg-Ni基合金. ...
Progress of hydrogen storage alloys for Ni-MH rechargeable power batteries in electric vehicles: A review
3
2017
... AB3型的LaMg2Ni9合金结构之一为如图4[52]所示的PuNi3型结构[53],其可看成是AB5结构单元(CaCu5型)和AB2结构单元(MgZn2型或MgCu2型)沿c轴堆叠而成.A2B7型RE-Mg-Ni系合金是在AB3型合金基础上发展起来的,只需改变沿c轴堆叠AB2单元和AB5结构单元比例即可,即A2B4 + 2 × AB5 = 2 × A2B7.由于Mg在RE-Mg-Ni基合金中固溶度有限,以及在合金中具有选择性占位,对合金的相组成有比较大的影响.一般来说,Mg原子只进入[A2B4]结构单元而不占据[AB5]单元.AB3型和A2B7型合金都具有比AB5合金更高的理论储氢量和对应的电化学容量[50,54].Kohno等[55]研究了La-Mg-Nix(x = 3~3.5)系列合金的电化学性能,发现x = 3.0、3.3和3.5的3种合金电极对应的放电容量分别为387、410和390 mAh/g,均高于LaNi5合金电极 320 mAh/g的容量.RE-Mg-Ni系合金高容量的优点,使之成为新一代Ni-MH电池负极材料.Pan等[56]研究了不同化学计量比La0.7Mg0.3(Ni0.85Co0.15)x(x = 2.5、3.0、3.5、4.0、5.0)合金的电化学性能,发现放电容量、高倍率放电性能、交换电流密度等均随x增加表现出先升后降的趋势.与之相对应的是,AB3主相的含量随x的增加而减少,AB5相含量恰恰相反.当x = 3.5时,合金由AB3主相和少量AB5相构成,这种多相结构合金电极的电化学性能最佳.该结果为后续开展多相复合以及多元合金化提供了重要启示. ...
... [
52]
(a) interrelation of stacking layers ...
... (b) some of the interstitial sites
Crystal structure of <i>AB</i><sub>3</sub>-type alloys<sup>[<xref ref-type="bibr" rid="R52">52</xref>]</sup>Fig.4![]()
A2B7型RE-Mg-Ni系合金中各元素对其电化学性能影响很大.添加Mg除了可以增加La2Ni7型合金的可逆吸/放氢能力,还将使其氢化过程由“各向异性”膨胀转变为“各向同性”膨胀,缓解了体积膨胀,从而稳定了晶格,抑制了合金在充/放电循环过程中的非晶化和歧化(disproportionation)[57].一般而言,A侧主要采用La、Ce或混合稀土取代价格相对较贵的Pr、Nd稀土.Pan等[58]采用Ce元素取代部分La,随着Ce含量的增加,La(La, Mg)2Ni9相丰度(phase abundance)增大,而LaNi5相丰度减小.与之对应的是,随着Ce含量的增加,合金的放电容量降低,但循环稳定性逐渐提高,这主要得益于Ce取代后电极合金晶胞参数的减小从而减小了体积膨胀.B侧取代元素种类繁多,其中比较关键的替代元素为Mn、Co和Al.适量的Mn取代可以提高A2B7型RE-Mg-Ni系合金的最大放电容量,改善合金电极的电化学动力学性能,但对循环稳定性影响不大[59,60].金属Co取代部分Ni后,降低了合金氢化后的体积膨胀,使合金在循环过程中体积变化较小.同时Co取代在合金表面形成高电催化活性的Ni-Co膜,有效提高了合金的电化学倍率性能,改善了A2B7型合金的循环稳定性[61].但Co的添加也会导致最大放电容量的衰减和成本增加,过高的Co替代使得氢扩散速率减小,导致合金高倍率放电(HRD)能力下降[61~63].Al替代Ni会导致合金表面形成致密的氧化膜,可以显著降低电池的腐蚀速率,减轻电极电化学反应过程中的体积膨胀和收缩进而提高电极的抗粉化能力[64].Chu等[65]研究了La0.7Mg0.3Ni3.5-x(MnAl)x(x = 0~0.2)的电化学性能,当x < 0.1时,合金电极的电化学反应动力学随Al含量的提高而降低,这主要是由于Al钝化膜的形成;当x > 0.1时,合金电极的电化学动力学加快,这主要由Mn溶解到碱性电解液中导致Al氧化膜破裂造成的.Liu等[66]在元素替代方面开展了大量的工作并对其进行了总结.为了进一步提高合金电极的循环稳定性和降低电极成本,Cao等[67]发明了高Sm低Co的La0.95Sm0.66Mg0.40Ni6.25-Al0.42Co0.32合金,其在2C和5C (1C是指1倍电池容量下电池在1 h完全充/放电时的电流强度)电流密度下放电容量分别为311.8和227.0 mAh/g,1C下经过239 cyc循环后容量保持率为80%,表现出较高的放电能力和优良的循环稳定性能.为进一步降低合金电极的成本,Ouyang等[68]制备了高Sm,无Pr、Nd、Co,由La2Ni7和LaNi5 2相组成的A2B7型La13.9Sm24.7-Mg1.5Ni58Al1.7Zr0.14Ag0.07合金,233 K下的脱氢平台压为0.005 MPa,低温放电和HRD能力强.如图5[68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
Synthesis and structure determination of a new series of hydrogen storage alloys; RMg2Ni9 (R = La, Ce, Pr, Nd, Sm and Gd) built from MgNi2 laves-type layers alternating with AB5 layers
1
1997
... AB3型的LaMg2Ni9合金结构之一为如图4[52]所示的PuNi3型结构[53],其可看成是AB5结构单元(CaCu5型)和AB2结构单元(MgZn2型或MgCu2型)沿c轴堆叠而成.A2B7型RE-Mg-Ni系合金是在AB3型合金基础上发展起来的,只需改变沿c轴堆叠AB2单元和AB5结构单元比例即可,即A2B4 + 2 × AB5 = 2 × A2B7.由于Mg在RE-Mg-Ni基合金中固溶度有限,以及在合金中具有选择性占位,对合金的相组成有比较大的影响.一般来说,Mg原子只进入[A2B4]结构单元而不占据[AB5]单元.AB3型和A2B7型合金都具有比AB5合金更高的理论储氢量和对应的电化学容量[50,54].Kohno等[55]研究了La-Mg-Nix(x = 3~3.5)系列合金的电化学性能,发现x = 3.0、3.3和3.5的3种合金电极对应的放电容量分别为387、410和390 mAh/g,均高于LaNi5合金电极 320 mAh/g的容量.RE-Mg-Ni系合金高容量的优点,使之成为新一代Ni-MH电池负极材料.Pan等[56]研究了不同化学计量比La0.7Mg0.3(Ni0.85Co0.15)x(x = 2.5、3.0、3.5、4.0、5.0)合金的电化学性能,发现放电容量、高倍率放电性能、交换电流密度等均随x增加表现出先升后降的趋势.与之相对应的是,AB3主相的含量随x的增加而减少,AB5相含量恰恰相反.当x = 3.5时,合金由AB3主相和少量AB5相构成,这种多相结构合金电极的电化学性能最佳.该结果为后续开展多相复合以及多元合金化提供了重要启示. ...
Microstructure and hydrogen storage properties of a multi-phase Ml0.7Mg0.3Ni3.2 hydrogen storage alloy
1
2004
... AB3型的LaMg2Ni9合金结构之一为如图4[52]所示的PuNi3型结构[53],其可看成是AB5结构单元(CaCu5型)和AB2结构单元(MgZn2型或MgCu2型)沿c轴堆叠而成.A2B7型RE-Mg-Ni系合金是在AB3型合金基础上发展起来的,只需改变沿c轴堆叠AB2单元和AB5结构单元比例即可,即A2B4 + 2 × AB5 = 2 × A2B7.由于Mg在RE-Mg-Ni基合金中固溶度有限,以及在合金中具有选择性占位,对合金的相组成有比较大的影响.一般来说,Mg原子只进入[A2B4]结构单元而不占据[AB5]单元.AB3型和A2B7型合金都具有比AB5合金更高的理论储氢量和对应的电化学容量[50,54].Kohno等[55]研究了La-Mg-Nix(x = 3~3.5)系列合金的电化学性能,发现x = 3.0、3.3和3.5的3种合金电极对应的放电容量分别为387、410和390 mAh/g,均高于LaNi5合金电极 320 mAh/g的容量.RE-Mg-Ni系合金高容量的优点,使之成为新一代Ni-MH电池负极材料.Pan等[56]研究了不同化学计量比La0.7Mg0.3(Ni0.85Co0.15)x(x = 2.5、3.0、3.5、4.0、5.0)合金的电化学性能,发现放电容量、高倍率放电性能、交换电流密度等均随x增加表现出先升后降的趋势.与之相对应的是,AB3主相的含量随x的增加而减少,AB5相含量恰恰相反.当x = 3.5时,合金由AB3主相和少量AB5相构成,这种多相结构合金电极的电化学性能最佳.该结果为后续开展多相复合以及多元合金化提供了重要启示. ...
Hydrogen storage properties of new ternary system alloys: La2MgNi9, La5Mg2Ni23, La3MgNi14
1
2000
... AB3型的LaMg2Ni9合金结构之一为如图4[52]所示的PuNi3型结构[53],其可看成是AB5结构单元(CaCu5型)和AB2结构单元(MgZn2型或MgCu2型)沿c轴堆叠而成.A2B7型RE-Mg-Ni系合金是在AB3型合金基础上发展起来的,只需改变沿c轴堆叠AB2单元和AB5结构单元比例即可,即A2B4 + 2 × AB5 = 2 × A2B7.由于Mg在RE-Mg-Ni基合金中固溶度有限,以及在合金中具有选择性占位,对合金的相组成有比较大的影响.一般来说,Mg原子只进入[A2B4]结构单元而不占据[AB5]单元.AB3型和A2B7型合金都具有比AB5合金更高的理论储氢量和对应的电化学容量[50,54].Kohno等[55]研究了La-Mg-Nix(x = 3~3.5)系列合金的电化学性能,发现x = 3.0、3.3和3.5的3种合金电极对应的放电容量分别为387、410和390 mAh/g,均高于LaNi5合金电极 320 mAh/g的容量.RE-Mg-Ni系合金高容量的优点,使之成为新一代Ni-MH电池负极材料.Pan等[56]研究了不同化学计量比La0.7Mg0.3(Ni0.85Co0.15)x(x = 2.5、3.0、3.5、4.0、5.0)合金的电化学性能,发现放电容量、高倍率放电性能、交换电流密度等均随x增加表现出先升后降的趋势.与之相对应的是,AB3主相的含量随x的增加而减少,AB5相含量恰恰相反.当x = 3.5时,合金由AB3主相和少量AB5相构成,这种多相结构合金电极的电化学性能最佳.该结果为后续开展多相复合以及多元合金化提供了重要启示. ...
A study of the structural and electrochemical properties of La0.7Mg0.3(Ni0.85Co0.15)x (x = 2.5-5.0) hydrogen storage alloys
1
2003
... AB3型的LaMg2Ni9合金结构之一为如图4[52]所示的PuNi3型结构[53],其可看成是AB5结构单元(CaCu5型)和AB2结构单元(MgZn2型或MgCu2型)沿c轴堆叠而成.A2B7型RE-Mg-Ni系合金是在AB3型合金基础上发展起来的,只需改变沿c轴堆叠AB2单元和AB5结构单元比例即可,即A2B4 + 2 × AB5 = 2 × A2B7.由于Mg在RE-Mg-Ni基合金中固溶度有限,以及在合金中具有选择性占位,对合金的相组成有比较大的影响.一般来说,Mg原子只进入[A2B4]结构单元而不占据[AB5]单元.AB3型和A2B7型合金都具有比AB5合金更高的理论储氢量和对应的电化学容量[50,54].Kohno等[55]研究了La-Mg-Nix(x = 3~3.5)系列合金的电化学性能,发现x = 3.0、3.3和3.5的3种合金电极对应的放电容量分别为387、410和390 mAh/g,均高于LaNi5合金电极 320 mAh/g的容量.RE-Mg-Ni系合金高容量的优点,使之成为新一代Ni-MH电池负极材料.Pan等[56]研究了不同化学计量比La0.7Mg0.3(Ni0.85Co0.15)x(x = 2.5、3.0、3.5、4.0、5.0)合金的电化学性能,发现放电容量、高倍率放电性能、交换电流密度等均随x增加表现出先升后降的趋势.与之相对应的是,AB3主相的含量随x的增加而减少,AB5相含量恰恰相反.当x = 3.5时,合金由AB3主相和少量AB5相构成,这种多相结构合金电极的电化学性能最佳.该结果为后续开展多相复合以及多元合金化提供了重要启示. ...
Mg substitution effect on the hydrogenation behaviour, thermodynamic and structural properties of the La2Ni7-H(D)2 system
1
2008
... A2B7型RE-Mg-Ni系合金中各元素对其电化学性能影响很大.添加Mg除了可以增加La2Ni7型合金的可逆吸/放氢能力,还将使其氢化过程由“各向异性”膨胀转变为“各向同性”膨胀,缓解了体积膨胀,从而稳定了晶格,抑制了合金在充/放电循环过程中的非晶化和歧化(disproportionation)[57].一般而言,A侧主要采用La、Ce或混合稀土取代价格相对较贵的Pr、Nd稀土.Pan等[58]采用Ce元素取代部分La,随着Ce含量的增加,La(La, Mg)2Ni9相丰度(phase abundance)增大,而LaNi5相丰度减小.与之对应的是,随着Ce含量的增加,合金的放电容量降低,但循环稳定性逐渐提高,这主要得益于Ce取代后电极合金晶胞参数的减小从而减小了体积膨胀.B侧取代元素种类繁多,其中比较关键的替代元素为Mn、Co和Al.适量的Mn取代可以提高A2B7型RE-Mg-Ni系合金的最大放电容量,改善合金电极的电化学动力学性能,但对循环稳定性影响不大[59,60].金属Co取代部分Ni后,降低了合金氢化后的体积膨胀,使合金在循环过程中体积变化较小.同时Co取代在合金表面形成高电催化活性的Ni-Co膜,有效提高了合金的电化学倍率性能,改善了A2B7型合金的循环稳定性[61].但Co的添加也会导致最大放电容量的衰减和成本增加,过高的Co替代使得氢扩散速率减小,导致合金高倍率放电(HRD)能力下降[61~63].Al替代Ni会导致合金表面形成致密的氧化膜,可以显著降低电池的腐蚀速率,减轻电极电化学反应过程中的体积膨胀和收缩进而提高电极的抗粉化能力[64].Chu等[65]研究了La0.7Mg0.3Ni3.5-x(MnAl)x(x = 0~0.2)的电化学性能,当x < 0.1时,合金电极的电化学反应动力学随Al含量的提高而降低,这主要是由于Al钝化膜的形成;当x > 0.1时,合金电极的电化学动力学加快,这主要由Mn溶解到碱性电解液中导致Al氧化膜破裂造成的.Liu等[66]在元素替代方面开展了大量的工作并对其进行了总结.为了进一步提高合金电极的循环稳定性和降低电极成本,Cao等[67]发明了高Sm低Co的La0.95Sm0.66Mg0.40Ni6.25-Al0.42Co0.32合金,其在2C和5C (1C是指1倍电池容量下电池在1 h完全充/放电时的电流强度)电流密度下放电容量分别为311.8和227.0 mAh/g,1C下经过239 cyc循环后容量保持率为80%,表现出较高的放电能力和优良的循环稳定性能.为进一步降低合金电极的成本,Ouyang等[68]制备了高Sm,无Pr、Nd、Co,由La2Ni7和LaNi5 2相组成的A2B7型La13.9Sm24.7-Mg1.5Ni58Al1.7Zr0.14Ag0.07合金,233 K下的脱氢平台压为0.005 MPa,低温放电和HRD能力强.如图5[68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
Effect of the cerium content on the structural and electrochemical properties of the La0.7-xCexMg0.3-Ni2.875Mn0.1Co0.525 (x = 0-0.5) hydrogen storage alloys
1
2004
... A2B7型RE-Mg-Ni系合金中各元素对其电化学性能影响很大.添加Mg除了可以增加La2Ni7型合金的可逆吸/放氢能力,还将使其氢化过程由“各向异性”膨胀转变为“各向同性”膨胀,缓解了体积膨胀,从而稳定了晶格,抑制了合金在充/放电循环过程中的非晶化和歧化(disproportionation)[57].一般而言,A侧主要采用La、Ce或混合稀土取代价格相对较贵的Pr、Nd稀土.Pan等[58]采用Ce元素取代部分La,随着Ce含量的增加,La(La, Mg)2Ni9相丰度(phase abundance)增大,而LaNi5相丰度减小.与之对应的是,随着Ce含量的增加,合金的放电容量降低,但循环稳定性逐渐提高,这主要得益于Ce取代后电极合金晶胞参数的减小从而减小了体积膨胀.B侧取代元素种类繁多,其中比较关键的替代元素为Mn、Co和Al.适量的Mn取代可以提高A2B7型RE-Mg-Ni系合金的最大放电容量,改善合金电极的电化学动力学性能,但对循环稳定性影响不大[59,60].金属Co取代部分Ni后,降低了合金氢化后的体积膨胀,使合金在循环过程中体积变化较小.同时Co取代在合金表面形成高电催化活性的Ni-Co膜,有效提高了合金的电化学倍率性能,改善了A2B7型合金的循环稳定性[61].但Co的添加也会导致最大放电容量的衰减和成本增加,过高的Co替代使得氢扩散速率减小,导致合金高倍率放电(HRD)能力下降[61~63].Al替代Ni会导致合金表面形成致密的氧化膜,可以显著降低电池的腐蚀速率,减轻电极电化学反应过程中的体积膨胀和收缩进而提高电极的抗粉化能力[64].Chu等[65]研究了La0.7Mg0.3Ni3.5-x(MnAl)x(x = 0~0.2)的电化学性能,当x < 0.1时,合金电极的电化学反应动力学随Al含量的提高而降低,这主要是由于Al钝化膜的形成;当x > 0.1时,合金电极的电化学动力学加快,这主要由Mn溶解到碱性电解液中导致Al氧化膜破裂造成的.Liu等[66]在元素替代方面开展了大量的工作并对其进行了总结.为了进一步提高合金电极的循环稳定性和降低电极成本,Cao等[67]发明了高Sm低Co的La0.95Sm0.66Mg0.40Ni6.25-Al0.42Co0.32合金,其在2C和5C (1C是指1倍电池容量下电池在1 h完全充/放电时的电流强度)电流密度下放电容量分别为311.8和227.0 mAh/g,1C下经过239 cyc循环后容量保持率为80%,表现出较高的放电能力和优良的循环稳定性能.为进一步降低合金电极的成本,Ouyang等[68]制备了高Sm,无Pr、Nd、Co,由La2Ni7和LaNi5 2相组成的A2B7型La13.9Sm24.7-Mg1.5Ni58Al1.7Zr0.14Ag0.07合金,233 K下的脱氢平台压为0.005 MPa,低温放电和HRD能力强.如图5[68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
Structural and electrochemical properties of the La0.7Mg0.3Ni2.975-xCo0.525Mnx hydrogen storage electrode alloys
1
2004
... A2B7型RE-Mg-Ni系合金中各元素对其电化学性能影响很大.添加Mg除了可以增加La2Ni7型合金的可逆吸/放氢能力,还将使其氢化过程由“各向异性”膨胀转变为“各向同性”膨胀,缓解了体积膨胀,从而稳定了晶格,抑制了合金在充/放电循环过程中的非晶化和歧化(disproportionation)[57].一般而言,A侧主要采用La、Ce或混合稀土取代价格相对较贵的Pr、Nd稀土.Pan等[58]采用Ce元素取代部分La,随着Ce含量的增加,La(La, Mg)2Ni9相丰度(phase abundance)增大,而LaNi5相丰度减小.与之对应的是,随着Ce含量的增加,合金的放电容量降低,但循环稳定性逐渐提高,这主要得益于Ce取代后电极合金晶胞参数的减小从而减小了体积膨胀.B侧取代元素种类繁多,其中比较关键的替代元素为Mn、Co和Al.适量的Mn取代可以提高A2B7型RE-Mg-Ni系合金的最大放电容量,改善合金电极的电化学动力学性能,但对循环稳定性影响不大[59,60].金属Co取代部分Ni后,降低了合金氢化后的体积膨胀,使合金在循环过程中体积变化较小.同时Co取代在合金表面形成高电催化活性的Ni-Co膜,有效提高了合金的电化学倍率性能,改善了A2B7型合金的循环稳定性[61].但Co的添加也会导致最大放电容量的衰减和成本增加,过高的Co替代使得氢扩散速率减小,导致合金高倍率放电(HRD)能力下降[61~63].Al替代Ni会导致合金表面形成致密的氧化膜,可以显著降低电池的腐蚀速率,减轻电极电化学反应过程中的体积膨胀和收缩进而提高电极的抗粉化能力[64].Chu等[65]研究了La0.7Mg0.3Ni3.5-x(MnAl)x(x = 0~0.2)的电化学性能,当x < 0.1时,合金电极的电化学反应动力学随Al含量的提高而降低,这主要是由于Al钝化膜的形成;当x > 0.1时,合金电极的电化学动力学加快,这主要由Mn溶解到碱性电解液中导致Al氧化膜破裂造成的.Liu等[66]在元素替代方面开展了大量的工作并对其进行了总结.为了进一步提高合金电极的循环稳定性和降低电极成本,Cao等[67]发明了高Sm低Co的La0.95Sm0.66Mg0.40Ni6.25-Al0.42Co0.32合金,其在2C和5C (1C是指1倍电池容量下电池在1 h完全充/放电时的电流强度)电流密度下放电容量分别为311.8和227.0 mAh/g,1C下经过239 cyc循环后容量保持率为80%,表现出较高的放电能力和优良的循环稳定性能.为进一步降低合金电极的成本,Ouyang等[68]制备了高Sm,无Pr、Nd、Co,由La2Ni7和LaNi5 2相组成的A2B7型La13.9Sm24.7-Mg1.5Ni58Al1.7Zr0.14Ag0.07合金,233 K下的脱氢平台压为0.005 MPa,低温放电和HRD能力强.如图5[68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
Effect of substituting Mn for Ni on the hydrogen storage and electrochemical properties of ReNi2.6-x-MnxCo0.9 alloys
1
2010
... A2B7型RE-Mg-Ni系合金中各元素对其电化学性能影响很大.添加Mg除了可以增加La2Ni7型合金的可逆吸/放氢能力,还将使其氢化过程由“各向异性”膨胀转变为“各向同性”膨胀,缓解了体积膨胀,从而稳定了晶格,抑制了合金在充/放电循环过程中的非晶化和歧化(disproportionation)[57].一般而言,A侧主要采用La、Ce或混合稀土取代价格相对较贵的Pr、Nd稀土.Pan等[58]采用Ce元素取代部分La,随着Ce含量的增加,La(La, Mg)2Ni9相丰度(phase abundance)增大,而LaNi5相丰度减小.与之对应的是,随着Ce含量的增加,合金的放电容量降低,但循环稳定性逐渐提高,这主要得益于Ce取代后电极合金晶胞参数的减小从而减小了体积膨胀.B侧取代元素种类繁多,其中比较关键的替代元素为Mn、Co和Al.适量的Mn取代可以提高A2B7型RE-Mg-Ni系合金的最大放电容量,改善合金电极的电化学动力学性能,但对循环稳定性影响不大[59,60].金属Co取代部分Ni后,降低了合金氢化后的体积膨胀,使合金在循环过程中体积变化较小.同时Co取代在合金表面形成高电催化活性的Ni-Co膜,有效提高了合金的电化学倍率性能,改善了A2B7型合金的循环稳定性[61].但Co的添加也会导致最大放电容量的衰减和成本增加,过高的Co替代使得氢扩散速率减小,导致合金高倍率放电(HRD)能力下降[61~63].Al替代Ni会导致合金表面形成致密的氧化膜,可以显著降低电池的腐蚀速率,减轻电极电化学反应过程中的体积膨胀和收缩进而提高电极的抗粉化能力[64].Chu等[65]研究了La0.7Mg0.3Ni3.5-x(MnAl)x(x = 0~0.2)的电化学性能,当x < 0.1时,合金电极的电化学反应动力学随Al含量的提高而降低,这主要是由于Al钝化膜的形成;当x > 0.1时,合金电极的电化学动力学加快,这主要由Mn溶解到碱性电解液中导致Al氧化膜破裂造成的.Liu等[66]在元素替代方面开展了大量的工作并对其进行了总结.为了进一步提高合金电极的循环稳定性和降低电极成本,Cao等[67]发明了高Sm低Co的La0.95Sm0.66Mg0.40Ni6.25-Al0.42Co0.32合金,其在2C和5C (1C是指1倍电池容量下电池在1 h完全充/放电时的电流强度)电流密度下放电容量分别为311.8和227.0 mAh/g,1C下经过239 cyc循环后容量保持率为80%,表现出较高的放电能力和优良的循环稳定性能.为进一步降低合金电极的成本,Ouyang等[68]制备了高Sm,无Pr、Nd、Co,由La2Ni7和LaNi5 2相组成的A2B7型La13.9Sm24.7-Mg1.5Ni58Al1.7Zr0.14Ag0.07合金,233 K下的脱氢平台压为0.005 MPa,低温放电和HRD能力强.如图5[68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
Effect of Co content on the structural and electrochemical properties of the La0.7Mg0.3Ni3.4-x-Mn0.1Cox hydride alloys: Ⅱ. Electrochemical properties
2
2004
... A2B7型RE-Mg-Ni系合金中各元素对其电化学性能影响很大.添加Mg除了可以增加La2Ni7型合金的可逆吸/放氢能力,还将使其氢化过程由“各向异性”膨胀转变为“各向同性”膨胀,缓解了体积膨胀,从而稳定了晶格,抑制了合金在充/放电循环过程中的非晶化和歧化(disproportionation)[57].一般而言,A侧主要采用La、Ce或混合稀土取代价格相对较贵的Pr、Nd稀土.Pan等[58]采用Ce元素取代部分La,随着Ce含量的增加,La(La, Mg)2Ni9相丰度(phase abundance)增大,而LaNi5相丰度减小.与之对应的是,随着Ce含量的增加,合金的放电容量降低,但循环稳定性逐渐提高,这主要得益于Ce取代后电极合金晶胞参数的减小从而减小了体积膨胀.B侧取代元素种类繁多,其中比较关键的替代元素为Mn、Co和Al.适量的Mn取代可以提高A2B7型RE-Mg-Ni系合金的最大放电容量,改善合金电极的电化学动力学性能,但对循环稳定性影响不大[59,60].金属Co取代部分Ni后,降低了合金氢化后的体积膨胀,使合金在循环过程中体积变化较小.同时Co取代在合金表面形成高电催化活性的Ni-Co膜,有效提高了合金的电化学倍率性能,改善了A2B7型合金的循环稳定性[61].但Co的添加也会导致最大放电容量的衰减和成本增加,过高的Co替代使得氢扩散速率减小,导致合金高倍率放电(HRD)能力下降[61~63].Al替代Ni会导致合金表面形成致密的氧化膜,可以显著降低电池的腐蚀速率,减轻电极电化学反应过程中的体积膨胀和收缩进而提高电极的抗粉化能力[64].Chu等[65]研究了La0.7Mg0.3Ni3.5-x(MnAl)x(x = 0~0.2)的电化学性能,当x < 0.1时,合金电极的电化学反应动力学随Al含量的提高而降低,这主要是由于Al钝化膜的形成;当x > 0.1时,合金电极的电化学动力学加快,这主要由Mn溶解到碱性电解液中导致Al氧化膜破裂造成的.Liu等[66]在元素替代方面开展了大量的工作并对其进行了总结.为了进一步提高合金电极的循环稳定性和降低电极成本,Cao等[67]发明了高Sm低Co的La0.95Sm0.66Mg0.40Ni6.25-Al0.42Co0.32合金,其在2C和5C (1C是指1倍电池容量下电池在1 h完全充/放电时的电流强度)电流密度下放电容量分别为311.8和227.0 mAh/g,1C下经过239 cyc循环后容量保持率为80%,表现出较高的放电能力和优良的循环稳定性能.为进一步降低合金电极的成本,Ouyang等[68]制备了高Sm,无Pr、Nd、Co,由La2Ni7和LaNi5 2相组成的A2B7型La13.9Sm24.7-Mg1.5Ni58Al1.7Zr0.14Ag0.07合金,233 K下的脱氢平台压为0.005 MPa,低温放电和HRD能力强.如图5[68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
... [61~63].Al替代Ni会导致合金表面形成致密的氧化膜,可以显著降低电池的腐蚀速率,减轻电极电化学反应过程中的体积膨胀和收缩进而提高电极的抗粉化能力[64].Chu等[65]研究了La0.7Mg0.3Ni3.5-x(MnAl)x(x = 0~0.2)的电化学性能,当x < 0.1时,合金电极的电化学反应动力学随Al含量的提高而降低,这主要是由于Al钝化膜的形成;当x > 0.1时,合金电极的电化学动力学加快,这主要由Mn溶解到碱性电解液中导致Al氧化膜破裂造成的.Liu等[66]在元素替代方面开展了大量的工作并对其进行了总结.为了进一步提高合金电极的循环稳定性和降低电极成本,Cao等[67]发明了高Sm低Co的La0.95Sm0.66Mg0.40Ni6.25-Al0.42Co0.32合金,其在2C和5C (1C是指1倍电池容量下电池在1 h完全充/放电时的电流强度)电流密度下放电容量分别为311.8和227.0 mAh/g,1C下经过239 cyc循环后容量保持率为80%,表现出较高的放电能力和优良的循环稳定性能.为进一步降低合金电极的成本,Ouyang等[68]制备了高Sm,无Pr、Nd、Co,由La2Ni7和LaNi5 2相组成的A2B7型La13.9Sm24.7-Mg1.5Ni58Al1.7Zr0.14Ag0.07合金,233 K下的脱氢平台压为0.005 MPa,低温放电和HRD能力强.如图5[68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
Effect of Co content on the structure and electrochemical properties of La1.5Mg0.5Ni7-xCox (x = 0, 1.2, 1.8) hydrogen storage alloys
0
2006
Phase structure and electrochemical properties of La0.67Mg0.33Ni3.0-xCox (x = 0.0, 0.25, 0.5, 0.75) hydrogen storage alloys
1
2006
... A2B7型RE-Mg-Ni系合金中各元素对其电化学性能影响很大.添加Mg除了可以增加La2Ni7型合金的可逆吸/放氢能力,还将使其氢化过程由“各向异性”膨胀转变为“各向同性”膨胀,缓解了体积膨胀,从而稳定了晶格,抑制了合金在充/放电循环过程中的非晶化和歧化(disproportionation)[57].一般而言,A侧主要采用La、Ce或混合稀土取代价格相对较贵的Pr、Nd稀土.Pan等[58]采用Ce元素取代部分La,随着Ce含量的增加,La(La, Mg)2Ni9相丰度(phase abundance)增大,而LaNi5相丰度减小.与之对应的是,随着Ce含量的增加,合金的放电容量降低,但循环稳定性逐渐提高,这主要得益于Ce取代后电极合金晶胞参数的减小从而减小了体积膨胀.B侧取代元素种类繁多,其中比较关键的替代元素为Mn、Co和Al.适量的Mn取代可以提高A2B7型RE-Mg-Ni系合金的最大放电容量,改善合金电极的电化学动力学性能,但对循环稳定性影响不大[59,60].金属Co取代部分Ni后,降低了合金氢化后的体积膨胀,使合金在循环过程中体积变化较小.同时Co取代在合金表面形成高电催化活性的Ni-Co膜,有效提高了合金的电化学倍率性能,改善了A2B7型合金的循环稳定性[61].但Co的添加也会导致最大放电容量的衰减和成本增加,过高的Co替代使得氢扩散速率减小,导致合金高倍率放电(HRD)能力下降[61~63].Al替代Ni会导致合金表面形成致密的氧化膜,可以显著降低电池的腐蚀速率,减轻电极电化学反应过程中的体积膨胀和收缩进而提高电极的抗粉化能力[64].Chu等[65]研究了La0.7Mg0.3Ni3.5-x(MnAl)x(x = 0~0.2)的电化学性能,当x < 0.1时,合金电极的电化学反应动力学随Al含量的提高而降低,这主要是由于Al钝化膜的形成;当x > 0.1时,合金电极的电化学动力学加快,这主要由Mn溶解到碱性电解液中导致Al氧化膜破裂造成的.Liu等[66]在元素替代方面开展了大量的工作并对其进行了总结.为了进一步提高合金电极的循环稳定性和降低电极成本,Cao等[67]发明了高Sm低Co的La0.95Sm0.66Mg0.40Ni6.25-Al0.42Co0.32合金,其在2C和5C (1C是指1倍电池容量下电池在1 h完全充/放电时的电流强度)电流密度下放电容量分别为311.8和227.0 mAh/g,1C下经过239 cyc循环后容量保持率为80%,表现出较高的放电能力和优良的循环稳定性能.为进一步降低合金电极的成本,Ouyang等[68]制备了高Sm,无Pr、Nd、Co,由La2Ni7和LaNi5 2相组成的A2B7型La13.9Sm24.7-Mg1.5Ni58Al1.7Zr0.14Ag0.07合金,233 K下的脱氢平台压为0.005 MPa,低温放电和HRD能力强.如图5[68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
Electrochemical properties of the La0.7Mg0.3Ni2.65-xMn0.1Co0.75Alx (x = 0-0.5) hydrogen storage alloy electrodes
1
2005
... A2B7型RE-Mg-Ni系合金中各元素对其电化学性能影响很大.添加Mg除了可以增加La2Ni7型合金的可逆吸/放氢能力,还将使其氢化过程由“各向异性”膨胀转变为“各向同性”膨胀,缓解了体积膨胀,从而稳定了晶格,抑制了合金在充/放电循环过程中的非晶化和歧化(disproportionation)[57].一般而言,A侧主要采用La、Ce或混合稀土取代价格相对较贵的Pr、Nd稀土.Pan等[58]采用Ce元素取代部分La,随着Ce含量的增加,La(La, Mg)2Ni9相丰度(phase abundance)增大,而LaNi5相丰度减小.与之对应的是,随着Ce含量的增加,合金的放电容量降低,但循环稳定性逐渐提高,这主要得益于Ce取代后电极合金晶胞参数的减小从而减小了体积膨胀.B侧取代元素种类繁多,其中比较关键的替代元素为Mn、Co和Al.适量的Mn取代可以提高A2B7型RE-Mg-Ni系合金的最大放电容量,改善合金电极的电化学动力学性能,但对循环稳定性影响不大[59,60].金属Co取代部分Ni后,降低了合金氢化后的体积膨胀,使合金在循环过程中体积变化较小.同时Co取代在合金表面形成高电催化活性的Ni-Co膜,有效提高了合金的电化学倍率性能,改善了A2B7型合金的循环稳定性[61].但Co的添加也会导致最大放电容量的衰减和成本增加,过高的Co替代使得氢扩散速率减小,导致合金高倍率放电(HRD)能力下降[61~63].Al替代Ni会导致合金表面形成致密的氧化膜,可以显著降低电池的腐蚀速率,减轻电极电化学反应过程中的体积膨胀和收缩进而提高电极的抗粉化能力[64].Chu等[65]研究了La0.7Mg0.3Ni3.5-x(MnAl)x(x = 0~0.2)的电化学性能,当x < 0.1时,合金电极的电化学反应动力学随Al含量的提高而降低,这主要是由于Al钝化膜的形成;当x > 0.1时,合金电极的电化学动力学加快,这主要由Mn溶解到碱性电解液中导致Al氧化膜破裂造成的.Liu等[66]在元素替代方面开展了大量的工作并对其进行了总结.为了进一步提高合金电极的循环稳定性和降低电极成本,Cao等[67]发明了高Sm低Co的La0.95Sm0.66Mg0.40Ni6.25-Al0.42Co0.32合金,其在2C和5C (1C是指1倍电池容量下电池在1 h完全充/放电时的电流强度)电流密度下放电容量分别为311.8和227.0 mAh/g,1C下经过239 cyc循环后容量保持率为80%,表现出较高的放电能力和优良的循环稳定性能.为进一步降低合金电极的成本,Ouyang等[68]制备了高Sm,无Pr、Nd、Co,由La2Ni7和LaNi5 2相组成的A2B7型La13.9Sm24.7-Mg1.5Ni58Al1.7Zr0.14Ag0.07合金,233 K下的脱氢平台压为0.005 MPa,低温放电和HRD能力强.如图5[68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
Electrochemical performances of cobalt-free La0.7Mg0.3Ni3.5-x(MnAl2)x (x = 0-0.20) hydrogen storage alloy electrodes
1
2008
... A2B7型RE-Mg-Ni系合金中各元素对其电化学性能影响很大.添加Mg除了可以增加La2Ni7型合金的可逆吸/放氢能力,还将使其氢化过程由“各向异性”膨胀转变为“各向同性”膨胀,缓解了体积膨胀,从而稳定了晶格,抑制了合金在充/放电循环过程中的非晶化和歧化(disproportionation)[57].一般而言,A侧主要采用La、Ce或混合稀土取代价格相对较贵的Pr、Nd稀土.Pan等[58]采用Ce元素取代部分La,随着Ce含量的增加,La(La, Mg)2Ni9相丰度(phase abundance)增大,而LaNi5相丰度减小.与之对应的是,随着Ce含量的增加,合金的放电容量降低,但循环稳定性逐渐提高,这主要得益于Ce取代后电极合金晶胞参数的减小从而减小了体积膨胀.B侧取代元素种类繁多,其中比较关键的替代元素为Mn、Co和Al.适量的Mn取代可以提高A2B7型RE-Mg-Ni系合金的最大放电容量,改善合金电极的电化学动力学性能,但对循环稳定性影响不大[59,60].金属Co取代部分Ni后,降低了合金氢化后的体积膨胀,使合金在循环过程中体积变化较小.同时Co取代在合金表面形成高电催化活性的Ni-Co膜,有效提高了合金的电化学倍率性能,改善了A2B7型合金的循环稳定性[61].但Co的添加也会导致最大放电容量的衰减和成本增加,过高的Co替代使得氢扩散速率减小,导致合金高倍率放电(HRD)能力下降[61~63].Al替代Ni会导致合金表面形成致密的氧化膜,可以显著降低电池的腐蚀速率,减轻电极电化学反应过程中的体积膨胀和收缩进而提高电极的抗粉化能力[64].Chu等[65]研究了La0.7Mg0.3Ni3.5-x(MnAl)x(x = 0~0.2)的电化学性能,当x < 0.1时,合金电极的电化学反应动力学随Al含量的提高而降低,这主要是由于Al钝化膜的形成;当x > 0.1时,合金电极的电化学动力学加快,这主要由Mn溶解到碱性电解液中导致Al氧化膜破裂造成的.Liu等[66]在元素替代方面开展了大量的工作并对其进行了总结.为了进一步提高合金电极的循环稳定性和降低电极成本,Cao等[67]发明了高Sm低Co的La0.95Sm0.66Mg0.40Ni6.25-Al0.42Co0.32合金,其在2C和5C (1C是指1倍电池容量下电池在1 h完全充/放电时的电流强度)电流密度下放电容量分别为311.8和227.0 mAh/g,1C下经过239 cyc循环后容量保持率为80%,表现出较高的放电能力和优良的循环稳定性能.为进一步降低合金电极的成本,Ouyang等[68]制备了高Sm,无Pr、Nd、Co,由La2Ni7和LaNi5 2相组成的A2B7型La13.9Sm24.7-Mg1.5Ni58Al1.7Zr0.14Ag0.07合金,233 K下的脱氢平台压为0.005 MPa,低温放电和HRD能力强.如图5[68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
Rare earth-Mg-Ni-based hydrogen storage alloys as negative electrode materials for Ni/MH batteries
1
2011
... A2B7型RE-Mg-Ni系合金中各元素对其电化学性能影响很大.添加Mg除了可以增加La2Ni7型合金的可逆吸/放氢能力,还将使其氢化过程由“各向异性”膨胀转变为“各向同性”膨胀,缓解了体积膨胀,从而稳定了晶格,抑制了合金在充/放电循环过程中的非晶化和歧化(disproportionation)[57].一般而言,A侧主要采用La、Ce或混合稀土取代价格相对较贵的Pr、Nd稀土.Pan等[58]采用Ce元素取代部分La,随着Ce含量的增加,La(La, Mg)2Ni9相丰度(phase abundance)增大,而LaNi5相丰度减小.与之对应的是,随着Ce含量的增加,合金的放电容量降低,但循环稳定性逐渐提高,这主要得益于Ce取代后电极合金晶胞参数的减小从而减小了体积膨胀.B侧取代元素种类繁多,其中比较关键的替代元素为Mn、Co和Al.适量的Mn取代可以提高A2B7型RE-Mg-Ni系合金的最大放电容量,改善合金电极的电化学动力学性能,但对循环稳定性影响不大[59,60].金属Co取代部分Ni后,降低了合金氢化后的体积膨胀,使合金在循环过程中体积变化较小.同时Co取代在合金表面形成高电催化活性的Ni-Co膜,有效提高了合金的电化学倍率性能,改善了A2B7型合金的循环稳定性[61].但Co的添加也会导致最大放电容量的衰减和成本增加,过高的Co替代使得氢扩散速率减小,导致合金高倍率放电(HRD)能力下降[61~63].Al替代Ni会导致合金表面形成致密的氧化膜,可以显著降低电池的腐蚀速率,减轻电极电化学反应过程中的体积膨胀和收缩进而提高电极的抗粉化能力[64].Chu等[65]研究了La0.7Mg0.3Ni3.5-x(MnAl)x(x = 0~0.2)的电化学性能,当x < 0.1时,合金电极的电化学反应动力学随Al含量的提高而降低,这主要是由于Al钝化膜的形成;当x > 0.1时,合金电极的电化学动力学加快,这主要由Mn溶解到碱性电解液中导致Al氧化膜破裂造成的.Liu等[66]在元素替代方面开展了大量的工作并对其进行了总结.为了进一步提高合金电极的循环稳定性和降低电极成本,Cao等[67]发明了高Sm低Co的La0.95Sm0.66Mg0.40Ni6.25-Al0.42Co0.32合金,其在2C和5C (1C是指1倍电池容量下电池在1 h完全充/放电时的电流强度)电流密度下放电容量分别为311.8和227.0 mAh/g,1C下经过239 cyc循环后容量保持率为80%,表现出较高的放电能力和优良的循环稳定性能.为进一步降低合金电极的成本,Ouyang等[68]制备了高Sm,无Pr、Nd、Co,由La2Ni7和LaNi5 2相组成的A2B7型La13.9Sm24.7-Mg1.5Ni58Al1.7Zr0.14Ag0.07合金,233 K下的脱氢平台压为0.005 MPa,低温放电和HRD能力强.如图5[68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
Enhanced discharge capacity and cycling properties in high-samarium, praseodymium/neodymium-free, and low-cobalt A2B7 electrode materials for nickel-metal hydride battery
1
2015
... A2B7型RE-Mg-Ni系合金中各元素对其电化学性能影响很大.添加Mg除了可以增加La2Ni7型合金的可逆吸/放氢能力,还将使其氢化过程由“各向异性”膨胀转变为“各向同性”膨胀,缓解了体积膨胀,从而稳定了晶格,抑制了合金在充/放电循环过程中的非晶化和歧化(disproportionation)[57].一般而言,A侧主要采用La、Ce或混合稀土取代价格相对较贵的Pr、Nd稀土.Pan等[58]采用Ce元素取代部分La,随着Ce含量的增加,La(La, Mg)2Ni9相丰度(phase abundance)增大,而LaNi5相丰度减小.与之对应的是,随着Ce含量的增加,合金的放电容量降低,但循环稳定性逐渐提高,这主要得益于Ce取代后电极合金晶胞参数的减小从而减小了体积膨胀.B侧取代元素种类繁多,其中比较关键的替代元素为Mn、Co和Al.适量的Mn取代可以提高A2B7型RE-Mg-Ni系合金的最大放电容量,改善合金电极的电化学动力学性能,但对循环稳定性影响不大[59,60].金属Co取代部分Ni后,降低了合金氢化后的体积膨胀,使合金在循环过程中体积变化较小.同时Co取代在合金表面形成高电催化活性的Ni-Co膜,有效提高了合金的电化学倍率性能,改善了A2B7型合金的循环稳定性[61].但Co的添加也会导致最大放电容量的衰减和成本增加,过高的Co替代使得氢扩散速率减小,导致合金高倍率放电(HRD)能力下降[61~63].Al替代Ni会导致合金表面形成致密的氧化膜,可以显著降低电池的腐蚀速率,减轻电极电化学反应过程中的体积膨胀和收缩进而提高电极的抗粉化能力[64].Chu等[65]研究了La0.7Mg0.3Ni3.5-x(MnAl)x(x = 0~0.2)的电化学性能,当x < 0.1时,合金电极的电化学反应动力学随Al含量的提高而降低,这主要是由于Al钝化膜的形成;当x > 0.1时,合金电极的电化学动力学加快,这主要由Mn溶解到碱性电解液中导致Al氧化膜破裂造成的.Liu等[66]在元素替代方面开展了大量的工作并对其进行了总结.为了进一步提高合金电极的循环稳定性和降低电极成本,Cao等[67]发明了高Sm低Co的La0.95Sm0.66Mg0.40Ni6.25-Al0.42Co0.32合金,其在2C和5C (1C是指1倍电池容量下电池在1 h完全充/放电时的电流强度)电流密度下放电容量分别为311.8和227.0 mAh/g,1C下经过239 cyc循环后容量保持率为80%,表现出较高的放电能力和优良的循环稳定性能.为进一步降低合金电极的成本,Ouyang等[68]制备了高Sm,无Pr、Nd、Co,由La2Ni7和LaNi5 2相组成的A2B7型La13.9Sm24.7-Mg1.5Ni58Al1.7Zr0.14Ag0.07合金,233 K下的脱氢平台压为0.005 MPa,低温放电和HRD能力强.如图5[68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
Hydrogen storage and electrochemical properties of Pr, Nd and Co-free La13.9Sm24.7Mg1.5Ni58-Al1.7Zr0.14Ag0.07 alloy as a nickel-metal hydride battery electrode
5
2018
... A2B7型RE-Mg-Ni系合金中各元素对其电化学性能影响很大.添加Mg除了可以增加La2Ni7型合金的可逆吸/放氢能力,还将使其氢化过程由“各向异性”膨胀转变为“各向同性”膨胀,缓解了体积膨胀,从而稳定了晶格,抑制了合金在充/放电循环过程中的非晶化和歧化(disproportionation)[57].一般而言,A侧主要采用La、Ce或混合稀土取代价格相对较贵的Pr、Nd稀土.Pan等[58]采用Ce元素取代部分La,随着Ce含量的增加,La(La, Mg)2Ni9相丰度(phase abundance)增大,而LaNi5相丰度减小.与之对应的是,随着Ce含量的增加,合金的放电容量降低,但循环稳定性逐渐提高,这主要得益于Ce取代后电极合金晶胞参数的减小从而减小了体积膨胀.B侧取代元素种类繁多,其中比较关键的替代元素为Mn、Co和Al.适量的Mn取代可以提高A2B7型RE-Mg-Ni系合金的最大放电容量,改善合金电极的电化学动力学性能,但对循环稳定性影响不大[59,60].金属Co取代部分Ni后,降低了合金氢化后的体积膨胀,使合金在循环过程中体积变化较小.同时Co取代在合金表面形成高电催化活性的Ni-Co膜,有效提高了合金的电化学倍率性能,改善了A2B7型合金的循环稳定性[61].但Co的添加也会导致最大放电容量的衰减和成本增加,过高的Co替代使得氢扩散速率减小,导致合金高倍率放电(HRD)能力下降[61~63].Al替代Ni会导致合金表面形成致密的氧化膜,可以显著降低电池的腐蚀速率,减轻电极电化学反应过程中的体积膨胀和收缩进而提高电极的抗粉化能力[64].Chu等[65]研究了La0.7Mg0.3Ni3.5-x(MnAl)x(x = 0~0.2)的电化学性能,当x < 0.1时,合金电极的电化学反应动力学随Al含量的提高而降低,这主要是由于Al钝化膜的形成;当x > 0.1时,合金电极的电化学动力学加快,这主要由Mn溶解到碱性电解液中导致Al氧化膜破裂造成的.Liu等[66]在元素替代方面开展了大量的工作并对其进行了总结.为了进一步提高合金电极的循环稳定性和降低电极成本,Cao等[67]发明了高Sm低Co的La0.95Sm0.66Mg0.40Ni6.25-Al0.42Co0.32合金,其在2C和5C (1C是指1倍电池容量下电池在1 h完全充/放电时的电流强度)电流密度下放电容量分别为311.8和227.0 mAh/g,1C下经过239 cyc循环后容量保持率为80%,表现出较高的放电能力和优良的循环稳定性能.为进一步降低合金电极的成本,Ouyang等[68]制备了高Sm,无Pr、Nd、Co,由La2Ni7和LaNi5 2相组成的A2B7型La13.9Sm24.7-Mg1.5Ni58Al1.7Zr0.14Ag0.07合金,233 K下的脱氢平台压为0.005 MPa,低温放电和HRD能力强.如图5[68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
... [68]所示,在330、970和1600 mAh/g的电流密度下,高倍率放电率分别为97%、86%和78%,而在1C下放电循环200 cyc的容量保持率为86.2%.该合金已成功实现产业化,应用于Ni-MH动力电池. ...
... [
68]
High rate discharge (HRD) at 298 K (a) and evolution of the discharge capacity with cycle number at 1 C (b) of the La<sub>13.9</sub>Sm<sub>24.7</sub>Mg<sub>1.5</sub>Ni<sub>58</sub>Al<sub>1.7</sub>Zr<sub>0.14</sub>Ag<sub>0.07</sub> alloy electrode<sup>[<xref ref-type="bibr" rid="R68">68</xref>]</sup>Fig.5![]() <strong>2.2 </strong>多相复合提高<strong><i>A</i><sub>2</sub><i>B</i><sub>7</sub></strong>型<strong>RE-Mg-Ni</strong>系电极合金的宽温电化学性能
<strong>2.2 </strong>多相复合提高<strong><i>A</i><sub>2</sub><i>B</i><sub>7</sub></strong>型<strong>RE-Mg-Ni</strong>系电极合金的宽温电化学性能Ni-MH电池储氢合金负极低温电化学性能差,使其难以在低温寒冷天气环境下正常工作,因此,合金电极的宽温电化学性能是亟待解决的问题.Liu等[69]探讨了-20~30℃范围内温度对La0.7Mg0.3Ni2.875-Co0.52Mn0.1合金电极电化学性能的影响,证明合金电极的最大放电容量和HRD均随着温度的降低而减小.在温度高于10℃时,H在电极中的扩散系数是影响电极放电能力的主要因素;而在低温环境下电荷转移速率是影响电极放电能力的关键因素.因此,改善电极合金的低温电化学性能需要对电极的动力学进行调控,提高电极合金的电荷转移速率和氢在电极合金基体中的扩散能力. ...
... [
68]
Fig.5![]() <strong>2.2 </strong>多相复合提高<strong><i>A</i><sub>2</sub><i>B</i><sub>7</sub></strong>型<strong>RE-Mg-Ni</strong>系电极合金的宽温电化学性能
<strong>2.2 </strong>多相复合提高<strong><i>A</i><sub>2</sub><i>B</i><sub>7</sub></strong>型<strong>RE-Mg-Ni</strong>系电极合金的宽温电化学性能Ni-MH电池储氢合金负极低温电化学性能差,使其难以在低温寒冷天气环境下正常工作,因此,合金电极的宽温电化学性能是亟待解决的问题.Liu等[69]探讨了-20~30℃范围内温度对La0.7Mg0.3Ni2.875-Co0.52Mn0.1合金电极电化学性能的影响,证明合金电极的最大放电容量和HRD均随着温度的降低而减小.在温度高于10℃时,H在电极中的扩散系数是影响电极放电能力的主要因素;而在低温环境下电荷转移速率是影响电极放电能力的关键因素.因此,改善电极合金的低温电化学性能需要对电极的动力学进行调控,提高电极合金的电荷转移速率和氢在电极合金基体中的扩散能力. ...
... 多相复合可以有效改善电极合金的低温放电性能.Ouyang等[68]制备了由丰度分别为94.3%和5.7%的La2Ni7和LaNi5相组成的A2B7型La13.9Sm24.7Mg1.5Ni58-Al1.7Zr0.14Ag0.07合金,该合金电极在不同的温度和放电电流密度下表现出相当宽的放电电压平台(0.5~1.0 V),并且具有优异的低温性能.当温度降至248 K时,合金电极仍然可以在0.2C和0.5C的电流密度下分别保持238和160 mAh/g的高放电容量.这主要是由于合金中La含量的降低导致金属氢化物的稳定性降低,从而增加了低温下合金电极的放电效率.Young等[70]研究了由AB2、AB3、A2B7、A5B19和AB5相构成的含Mn的A2B7型Mm19.3Mg3.9Ni72.8-xAl4.0Mnx(x = 2.3、4.7、7.0、9.3)合金的电化学性能,与单相AB5合金相比,具有多相结构的A2B7型合金在低温下(-30和-40℃)表现出更高的放电容量.Ni等[71]制备了Mm0.7Mg0.3Ni2.58Co0.5Mn0.3Al0.12合金,证明Mg的添加使合金生成了LaNi5和La2Ni7双相结构,其在-35、0和30℃条件下,合金的放电容量、循环稳定性、自放电和抗粉化性能均优于仅为单一LaNi5相的Mm0.7Ni2.58-Co0.5Mn0.3Al0.12合金.Shen等[72]研究了Nd含量对La0.8-x-NdxMg0.2Ni3.1Co0.25Al0.15合金低温(-40℃)电化学性能的影响,表明La0.7Nd0.1Mg0.2Ni3.1Co0.25Al0.15合金中具有更多的LaNi5催化相和(La, Mg)2Ni7相,且LaMgNi4杂质相更少,因此在低温下这种多相结构电极的电化学性能最佳,其放电容量取决于相应合金的相丰度和动力学,随着Nd含量的提高,在-40℃下电极合金的最大放电容量首先从188.5 mAh/g (x = 0)增加到201.7 mAh/g (x = 0.1),再降低至153.9 mAh/g (x = 0.4). ...
The electrochemical performance of a La-Mg-Ni-Co-Mn metal hydride electrode alloy in the temperature range of -20 to 30℃
1
2004
... Ni-MH电池储氢合金负极低温电化学性能差,使其难以在低温寒冷天气环境下正常工作,因此,合金电极的宽温电化学性能是亟待解决的问题.Liu等[69]探讨了-20~30℃范围内温度对La0.7Mg0.3Ni2.875-Co0.52Mn0.1合金电极电化学性能的影响,证明合金电极的最大放电容量和HRD均随着温度的降低而减小.在温度高于10℃时,H在电极中的扩散系数是影响电极放电能力的主要因素;而在低温环境下电荷转移速率是影响电极放电能力的关键因素.因此,改善电极合金的低温电化学性能需要对电极的动力学进行调控,提高电极合金的电荷转移速率和氢在电极合金基体中的扩散能力. ...
Mn in misch-metal based superlattice metal hydride alloy-Part 2 Ni/MH battery performance and failure mechanism
1
2015
... 多相复合可以有效改善电极合金的低温放电性能.Ouyang等[68]制备了由丰度分别为94.3%和5.7%的La2Ni7和LaNi5相组成的A2B7型La13.9Sm24.7Mg1.5Ni58-Al1.7Zr0.14Ag0.07合金,该合金电极在不同的温度和放电电流密度下表现出相当宽的放电电压平台(0.5~1.0 V),并且具有优异的低温性能.当温度降至248 K时,合金电极仍然可以在0.2C和0.5C的电流密度下分别保持238和160 mAh/g的高放电容量.这主要是由于合金中La含量的降低导致金属氢化物的稳定性降低,从而增加了低温下合金电极的放电效率.Young等[70]研究了由AB2、AB3、A2B7、A5B19和AB5相构成的含Mn的A2B7型Mm19.3Mg3.9Ni72.8-xAl4.0Mnx(x = 2.3、4.7、7.0、9.3)合金的电化学性能,与单相AB5合金相比,具有多相结构的A2B7型合金在低温下(-30和-40℃)表现出更高的放电容量.Ni等[71]制备了Mm0.7Mg0.3Ni2.58Co0.5Mn0.3Al0.12合金,证明Mg的添加使合金生成了LaNi5和La2Ni7双相结构,其在-35、0和30℃条件下,合金的放电容量、循环稳定性、自放电和抗粉化性能均优于仅为单一LaNi5相的Mm0.7Ni2.58-Co0.5Mn0.3Al0.12合金.Shen等[72]研究了Nd含量对La0.8-x-NdxMg0.2Ni3.1Co0.25Al0.15合金低温(-40℃)电化学性能的影响,表明La0.7Nd0.1Mg0.2Ni3.1Co0.25Al0.15合金中具有更多的LaNi5催化相和(La, Mg)2Ni7相,且LaMgNi4杂质相更少,因此在低温下这种多相结构电极的电化学性能最佳,其放电容量取决于相应合金的相丰度和动力学,随着Nd含量的提高,在-40℃下电极合金的最大放电容量首先从188.5 mAh/g (x = 0)增加到201.7 mAh/g (x = 0.1),再降低至153.9 mAh/g (x = 0.4). ...
Electrochemical performances of Mm0.7MgxNi2.58Co0.5Mn0.3Al0.12 (x = 0, 0.3) hydrogen storage alloys in the temperature range from 238 to 303 K
1
2012
... 多相复合可以有效改善电极合金的低温放电性能.Ouyang等[68]制备了由丰度分别为94.3%和5.7%的La2Ni7和LaNi5相组成的A2B7型La13.9Sm24.7Mg1.5Ni58-Al1.7Zr0.14Ag0.07合金,该合金电极在不同的温度和放电电流密度下表现出相当宽的放电电压平台(0.5~1.0 V),并且具有优异的低温性能.当温度降至248 K时,合金电极仍然可以在0.2C和0.5C的电流密度下分别保持238和160 mAh/g的高放电容量.这主要是由于合金中La含量的降低导致金属氢化物的稳定性降低,从而增加了低温下合金电极的放电效率.Young等[70]研究了由AB2、AB3、A2B7、A5B19和AB5相构成的含Mn的A2B7型Mm19.3Mg3.9Ni72.8-xAl4.0Mnx(x = 2.3、4.7、7.0、9.3)合金的电化学性能,与单相AB5合金相比,具有多相结构的A2B7型合金在低温下(-30和-40℃)表现出更高的放电容量.Ni等[71]制备了Mm0.7Mg0.3Ni2.58Co0.5Mn0.3Al0.12合金,证明Mg的添加使合金生成了LaNi5和La2Ni7双相结构,其在-35、0和30℃条件下,合金的放电容量、循环稳定性、自放电和抗粉化性能均优于仅为单一LaNi5相的Mm0.7Ni2.58-Co0.5Mn0.3Al0.12合金.Shen等[72]研究了Nd含量对La0.8-x-NdxMg0.2Ni3.1Co0.25Al0.15合金低温(-40℃)电化学性能的影响,表明La0.7Nd0.1Mg0.2Ni3.1Co0.25Al0.15合金中具有更多的LaNi5催化相和(La, Mg)2Ni7相,且LaMgNi4杂质相更少,因此在低温下这种多相结构电极的电化学性能最佳,其放电容量取决于相应合金的相丰度和动力学,随着Nd含量的提高,在-40℃下电极合金的最大放电容量首先从188.5 mAh/g (x = 0)增加到201.7 mAh/g (x = 0.1),再降低至153.9 mAh/g (x = 0.4). ...
The structure and 233 K electrochemical properties of La0.8-xNdxMg0.2Ni3.1Co0.25Al0.15 (x = 0.0-0.4) hydrogen storage alloys
1
2009
... 多相复合可以有效改善电极合金的低温放电性能.Ouyang等[68]制备了由丰度分别为94.3%和5.7%的La2Ni7和LaNi5相组成的A2B7型La13.9Sm24.7Mg1.5Ni58-Al1.7Zr0.14Ag0.07合金,该合金电极在不同的温度和放电电流密度下表现出相当宽的放电电压平台(0.5~1.0 V),并且具有优异的低温性能.当温度降至248 K时,合金电极仍然可以在0.2C和0.5C的电流密度下分别保持238和160 mAh/g的高放电容量.这主要是由于合金中La含量的降低导致金属氢化物的稳定性降低,从而增加了低温下合金电极的放电效率.Young等[70]研究了由AB2、AB3、A2B7、A5B19和AB5相构成的含Mn的A2B7型Mm19.3Mg3.9Ni72.8-xAl4.0Mnx(x = 2.3、4.7、7.0、9.3)合金的电化学性能,与单相AB5合金相比,具有多相结构的A2B7型合金在低温下(-30和-40℃)表现出更高的放电容量.Ni等[71]制备了Mm0.7Mg0.3Ni2.58Co0.5Mn0.3Al0.12合金,证明Mg的添加使合金生成了LaNi5和La2Ni7双相结构,其在-35、0和30℃条件下,合金的放电容量、循环稳定性、自放电和抗粉化性能均优于仅为单一LaNi5相的Mm0.7Ni2.58-Co0.5Mn0.3Al0.12合金.Shen等[72]研究了Nd含量对La0.8-x-NdxMg0.2Ni3.1Co0.25Al0.15合金低温(-40℃)电化学性能的影响,表明La0.7Nd0.1Mg0.2Ni3.1Co0.25Al0.15合金中具有更多的LaNi5催化相和(La, Mg)2Ni7相,且LaMgNi4杂质相更少,因此在低温下这种多相结构电极的电化学性能最佳,其放电容量取决于相应合金的相丰度和动力学,随着Nd含量的提高,在-40℃下电极合金的最大放电容量首先从188.5 mAh/g (x = 0)增加到201.7 mAh/g (x = 0.1),再降低至153.9 mAh/g (x = 0.4). ...
Electrochemical behaviour of some mechanically alloyed Mg-Ni-based amorphous hydrogen storage alloys
1
1994
... Lei等[73]发现球磨法制备的Mg-Ni非晶合金能够在室温下可逆地进行电化学吸/放氢,并有很高的容量.其后不少研究人员用球磨和薄膜技术制备了多种多元Mg-Ni基非晶合金,都获得了很高的电化学容量[74~77].遗憾的是,其循环稳定性极差.过去普遍认为,由于Mg自身腐蚀电位较低,镁基非晶合金充/放电循环过程中的容量衰减主要是其被碱液氧化腐蚀生成MgO或Mg(OH)2,以及循环过程中由于应力导致合金开裂和粉化而加剧腐蚀导致容量衰减[78~80].Lee等[81]使用XPS对机械合金化制备的(Mg1 - xZrx)2Ni (x = 0.1、0.3)合金进行分析,发现非晶态或纳米晶Mg表面原子的结合能要低于常规的晶态Mg,非晶或纳米晶电极的表面能较常规晶态电极亦有所增加,电化学测试结果表明H在电极合金中的扩散速率和电荷转移反应加快,但也加快了Mg(OH)2钝化层的形成和电极的迅速溶解.尽管研究人员采用元素替代、表面处理、多相复合等方法努力抑制腐蚀,但收效甚微.这也给我们启示,容量衰减可能还有其他原因. ...
High energy density strategies: From hydride-forming materials research to battery integration
1
2004
... Lei等[73]发现球磨法制备的Mg-Ni非晶合金能够在室温下可逆地进行电化学吸/放氢,并有很高的容量.其后不少研究人员用球磨和薄膜技术制备了多种多元Mg-Ni基非晶合金,都获得了很高的电化学容量[74~77].遗憾的是,其循环稳定性极差.过去普遍认为,由于Mg自身腐蚀电位较低,镁基非晶合金充/放电循环过程中的容量衰减主要是其被碱液氧化腐蚀生成MgO或Mg(OH)2,以及循环过程中由于应力导致合金开裂和粉化而加剧腐蚀导致容量衰减[78~80].Lee等[81]使用XPS对机械合金化制备的(Mg1 - xZrx)2Ni (x = 0.1、0.3)合金进行分析,发现非晶态或纳米晶Mg表面原子的结合能要低于常规的晶态Mg,非晶或纳米晶电极的表面能较常规晶态电极亦有所增加,电化学测试结果表明H在电极合金中的扩散速率和电荷转移反应加快,但也加快了Mg(OH)2钝化层的形成和电极的迅速溶解.尽管研究人员采用元素替代、表面处理、多相复合等方法努力抑制腐蚀,但收效甚微.这也给我们启示,容量衰减可能还有其他原因. ...
Structural and hydriding properties of the Mg-Ni-H system with nano- and/or amorphous structures
0
1997
Hydriding and dehydriding characteristics of an amorphous Mg2Ni-Ni composite
0
1999
The effect of partial substitution of Zr for Ti on the electrochemical properties and surface passivation film of Mg35Ti10-xZrxNi55 (x = 1, 3, 5, 7, 9) electrode alloys
1
2002
... Lei等[73]发现球磨法制备的Mg-Ni非晶合金能够在室温下可逆地进行电化学吸/放氢,并有很高的容量.其后不少研究人员用球磨和薄膜技术制备了多种多元Mg-Ni基非晶合金,都获得了很高的电化学容量[74~77].遗憾的是,其循环稳定性极差.过去普遍认为,由于Mg自身腐蚀电位较低,镁基非晶合金充/放电循环过程中的容量衰减主要是其被碱液氧化腐蚀生成MgO或Mg(OH)2,以及循环过程中由于应力导致合金开裂和粉化而加剧腐蚀导致容量衰减[78~80].Lee等[81]使用XPS对机械合金化制备的(Mg1 - xZrx)2Ni (x = 0.1、0.3)合金进行分析,发现非晶态或纳米晶Mg表面原子的结合能要低于常规的晶态Mg,非晶或纳米晶电极的表面能较常规晶态电极亦有所增加,电化学测试结果表明H在电极合金中的扩散速率和电荷转移反应加快,但也加快了Mg(OH)2钝化层的形成和电极的迅速溶解.尽管研究人员采用元素替代、表面处理、多相复合等方法努力抑制腐蚀,但收效甚微.这也给我们启示,容量衰减可能还有其他原因. ...
A study of the degradation of the electrochemical capacity of amorphous Mg50Ni50 alloy
1
1996
... Lei等[73]发现球磨法制备的Mg-Ni非晶合金能够在室温下可逆地进行电化学吸/放氢,并有很高的容量.其后不少研究人员用球磨和薄膜技术制备了多种多元Mg-Ni基非晶合金,都获得了很高的电化学容量[74~77].遗憾的是,其循环稳定性极差.过去普遍认为,由于Mg自身腐蚀电位较低,镁基非晶合金充/放电循环过程中的容量衰减主要是其被碱液氧化腐蚀生成MgO或Mg(OH)2,以及循环过程中由于应力导致合金开裂和粉化而加剧腐蚀导致容量衰减[78~80].Lee等[81]使用XPS对机械合金化制备的(Mg1 - xZrx)2Ni (x = 0.1、0.3)合金进行分析,发现非晶态或纳米晶Mg表面原子的结合能要低于常规的晶态Mg,非晶或纳米晶电极的表面能较常规晶态电极亦有所增加,电化学测试结果表明H在电极合金中的扩散速率和电荷转移反应加快,但也加快了Mg(OH)2钝化层的形成和电极的迅速溶解.尽管研究人员采用元素替代、表面处理、多相复合等方法努力抑制腐蚀,但收效甚微.这也给我们启示,容量衰减可能还有其他原因. ...
The effect of Ni content on the electrochemical and surface characteristics of Mg90-xTi10Nix (x = 50, 55, 60) ternary hydrogen storage electrode alloys
0
2001
Degradation kinetics of discharge capacity for amorphous Mg-Ni electrode
1
2002
... Lei等[73]发现球磨法制备的Mg-Ni非晶合金能够在室温下可逆地进行电化学吸/放氢,并有很高的容量.其后不少研究人员用球磨和薄膜技术制备了多种多元Mg-Ni基非晶合金,都获得了很高的电化学容量[74~77].遗憾的是,其循环稳定性极差.过去普遍认为,由于Mg自身腐蚀电位较低,镁基非晶合金充/放电循环过程中的容量衰减主要是其被碱液氧化腐蚀生成MgO或Mg(OH)2,以及循环过程中由于应力导致合金开裂和粉化而加剧腐蚀导致容量衰减[78~80].Lee等[81]使用XPS对机械合金化制备的(Mg1 - xZrx)2Ni (x = 0.1、0.3)合金进行分析,发现非晶态或纳米晶Mg表面原子的结合能要低于常规的晶态Mg,非晶或纳米晶电极的表面能较常规晶态电极亦有所增加,电化学测试结果表明H在电极合金中的扩散速率和电荷转移反应加快,但也加快了Mg(OH)2钝化层的形成和电极的迅速溶解.尽管研究人员采用元素替代、表面处理、多相复合等方法努力抑制腐蚀,但收效甚微.这也给我们启示,容量衰减可能还有其他原因. ...
The surface state of nanocrystalline and amorphous Mg2Ni alloys prepared by mechanical alloying
1
2000
... Lei等[73]发现球磨法制备的Mg-Ni非晶合金能够在室温下可逆地进行电化学吸/放氢,并有很高的容量.其后不少研究人员用球磨和薄膜技术制备了多种多元Mg-Ni基非晶合金,都获得了很高的电化学容量[74~77].遗憾的是,其循环稳定性极差.过去普遍认为,由于Mg自身腐蚀电位较低,镁基非晶合金充/放电循环过程中的容量衰减主要是其被碱液氧化腐蚀生成MgO或Mg(OH)2,以及循环过程中由于应力导致合金开裂和粉化而加剧腐蚀导致容量衰减[78~80].Lee等[81]使用XPS对机械合金化制备的(Mg1 - xZrx)2Ni (x = 0.1、0.3)合金进行分析,发现非晶态或纳米晶Mg表面原子的结合能要低于常规的晶态Mg,非晶或纳米晶电极的表面能较常规晶态电极亦有所增加,电化学测试结果表明H在电极合金中的扩散速率和电荷转移反应加快,但也加快了Mg(OH)2钝化层的形成和电极的迅速溶解.尽管研究人员采用元素替代、表面处理、多相复合等方法努力抑制腐蚀,但收效甚微.这也给我们启示,容量衰减可能还有其他原因. ...
Hydrogenation and crystallization of amorphous phase: A new mechanism for the electrochemical capacity and its decay in milled Mg-Ni alloys
5
2019
... Huang等[82]通过球磨10、30和60 h制备了不同结构特征的Mg0.5Ni0.5合金,合金中非晶相的含量随着球磨时间的延长而增加,相应的放电容量也明显提高.如图6a[82]所示,球磨Mg0.5Ni0.5合金电极的放电容量和合金中非晶相的含量成正比,Mg0.5Ni0.5合金在50 mAh/g的放电电流密度下,球磨10、30和60 h的放电容量分别为445、486和515 mAh/g,同时Mg-Ni合金放电容量保持率也随球磨时间的延长而提高,如图6b[82]所示.进一步的TEM、DSC等分析表明,合金容量衰减相当一部分归因于非晶相在充电吸氢过程中发生晶化而失去电化学放电能力.由此,证明非晶在放电过程的氢致晶化,也是镁基非晶电极合金在循环中容量衰减的重要原因. ...
... [82]所示,球磨Mg0.5Ni0.5合金电极的放电容量和合金中非晶相的含量成正比,Mg0.5Ni0.5合金在50 mAh/g的放电电流密度下,球磨10、30和60 h的放电容量分别为445、486和515 mAh/g,同时Mg-Ni合金放电容量保持率也随球磨时间的延长而提高,如图6b[82]所示.进一步的TEM、DSC等分析表明,合金容量衰减相当一部分归因于非晶相在充电吸氢过程中发生晶化而失去电化学放电能力.由此,证明非晶在放电过程的氢致晶化,也是镁基非晶电极合金在循环中容量衰减的重要原因. ...
... [82]所示.进一步的TEM、DSC等分析表明,合金容量衰减相当一部分归因于非晶相在充电吸氢过程中发生晶化而失去电化学放电能力.由此,证明非晶在放电过程的氢致晶化,也是镁基非晶电极合金在循环中容量衰减的重要原因. ...
... [
82]
The first charge/discharge curves (a) and the cycling performance (b) of the BM Mg<sub>0.5</sub>Ni<sub>0.5</sub> alloys<sup>[<xref ref-type="bibr" rid="R82">82</xref>]</sup>Fig.6![]() <strong>3.2 </strong>提高镁基非晶储氢合金循环稳定性的新路径探索
<strong>3.2 </strong>提高镁基非晶储氢合金循环稳定性的新路径探索非晶态Mg-Ni合金由于其独特的非晶结构促进了H在电极合金中的扩散以及电荷转移,但镁基非晶合金在强碱性电解液中耐腐蚀性较差,同时,放电中发生氢致晶化,导致其高放电容量在充/放电循环过程中迅速衰减,如何解决该问题是开发长寿命、高容量镁基非晶合金的关键. ...
... [
82]
Fig.6![]() <strong>3.2 </strong>提高镁基非晶储氢合金循环稳定性的新路径探索
<strong>3.2 </strong>提高镁基非晶储氢合金循环稳定性的新路径探索非晶态Mg-Ni合金由于其独特的非晶结构促进了H在电极合金中的扩散以及电荷转移,但镁基非晶合金在强碱性电解液中耐腐蚀性较差,同时,放电中发生氢致晶化,导致其高放电容量在充/放电循环过程中迅速衰减,如何解决该问题是开发长寿命、高容量镁基非晶合金的关键. ...
The capacity deterioration model of mechanically alloyed MgxNi100-x amorphous electrodes in charging-discharging cycling
1
1997
... 合金化常被用来改善镁基非晶合金电极的循环性能.Ni[83]、V[84]、Co[85]、Ti[86]等作为非晶镁基合金化的典型代表元素,可以有效改善非晶合金的电化学循环性能.Anik等[86]制备的Mg0.9Ti0.2Ni0.9合金电极在20 cyc充放电循环后,容量保持率高达72%,表明金属Ti合金化对电极的循环稳定性提高显著.过去认为Ti的加入使得充/放电过程中合金表面形成的TiO2层延缓了合金电极的腐蚀速率.Han等[87]对Mg0.7Ti0.3Ni1.0合金的电化学阻抗分析表明,在充/放电循环过程中Mg0.7Ti0.3Ni1.0合金与电解液之间的电荷转移电阻不会增加,Auger电子能谱(AES)也表明Mg0.7Ti0.3Ni1.0合金电极表面氧化层的厚度要小于MgNi合金电极表面的氧化层厚度.事实上,Ti的主要作用是提高了非晶的稳定性,避免了氢致非晶化.Huang等[88]在非晶Mg0.50Ni0.50合金中引入Ti制备了Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金,使晶化温度由Mg0.50Ni0.50合金的300℃提高到350℃.由图7a[88]可以看出,Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后TG曲线的失重率分别为17.3%、18.7%和17.0%,相近的失重率说明腐蚀速率基本相同,也说明Ti添加并未有效改善合金电极的耐腐蚀性,这意味着添加Ti对循环性能的改善另有原因.图7b[88]为球磨60 h制备的Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后的DSC曲线,Mg0.50Ni0.50的2个峰分别对应脱氢峰和Mg(OH)2分解的吸热峰,而添加了金属Ti的电极没有脱氢峰的出现,说明添加Ti可以使合金在充/放电的过程中保持可逆的吸/放氢反应.而且在370℃左右处Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50电极合金均出现了非晶相的晶化转变峰,也说明了添加了Ti的合金经30 cyc循环后非晶相依旧得到了保留,而Mg0.50Ni0.50电极合金则已经完全晶化.上述结果很好地说明了添加Ti可以抑制合金非晶相的氢致晶化转变,从而提高Mg-Ni非晶合金电极的稳定性.值得注意的是,添加Ti还导致了合金中形成TiNi催化相,从而提高了非晶合金电极的倍率性能和动力学性能,但随着Ti添加量的增加,TiNi相增多而非晶相减少,导致合金的放电容量降低.随后,Li等[89]通过球磨法探讨了添加In、Al、Cu和Sc等对Mg-Ni基非晶合金电化学循环稳定性和热力学稳定性的影响,发现添加Al、Cu和In可以抑制合金电极在充/放电循环中的氢致晶化,其中添加Al的作用最显著,其组成的合金电极具有更高的电化学容量保持率. ...
Electrochemical characteristics of an amorphous Mg0.9V0.1Ni alloy prepared by mechanical alloying
1
1998
... 合金化常被用来改善镁基非晶合金电极的循环性能.Ni[83]、V[84]、Co[85]、Ti[86]等作为非晶镁基合金化的典型代表元素,可以有效改善非晶合金的电化学循环性能.Anik等[86]制备的Mg0.9Ti0.2Ni0.9合金电极在20 cyc充放电循环后,容量保持率高达72%,表明金属Ti合金化对电极的循环稳定性提高显著.过去认为Ti的加入使得充/放电过程中合金表面形成的TiO2层延缓了合金电极的腐蚀速率.Han等[87]对Mg0.7Ti0.3Ni1.0合金的电化学阻抗分析表明,在充/放电循环过程中Mg0.7Ti0.3Ni1.0合金与电解液之间的电荷转移电阻不会增加,Auger电子能谱(AES)也表明Mg0.7Ti0.3Ni1.0合金电极表面氧化层的厚度要小于MgNi合金电极表面的氧化层厚度.事实上,Ti的主要作用是提高了非晶的稳定性,避免了氢致非晶化.Huang等[88]在非晶Mg0.50Ni0.50合金中引入Ti制备了Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金,使晶化温度由Mg0.50Ni0.50合金的300℃提高到350℃.由图7a[88]可以看出,Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后TG曲线的失重率分别为17.3%、18.7%和17.0%,相近的失重率说明腐蚀速率基本相同,也说明Ti添加并未有效改善合金电极的耐腐蚀性,这意味着添加Ti对循环性能的改善另有原因.图7b[88]为球磨60 h制备的Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后的DSC曲线,Mg0.50Ni0.50的2个峰分别对应脱氢峰和Mg(OH)2分解的吸热峰,而添加了金属Ti的电极没有脱氢峰的出现,说明添加Ti可以使合金在充/放电的过程中保持可逆的吸/放氢反应.而且在370℃左右处Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50电极合金均出现了非晶相的晶化转变峰,也说明了添加了Ti的合金经30 cyc循环后非晶相依旧得到了保留,而Mg0.50Ni0.50电极合金则已经完全晶化.上述结果很好地说明了添加Ti可以抑制合金非晶相的氢致晶化转变,从而提高Mg-Ni非晶合金电极的稳定性.值得注意的是,添加Ti还导致了合金中形成TiNi催化相,从而提高了非晶合金电极的倍率性能和动力学性能,但随着Ti添加量的增加,TiNi相增多而非晶相减少,导致合金的放电容量降低.随后,Li等[89]通过球磨法探讨了添加In、Al、Cu和Sc等对Mg-Ni基非晶合金电化学循环稳定性和热力学稳定性的影响,发现添加Al、Cu和In可以抑制合金电极在充/放电循环中的氢致晶化,其中添加Al的作用最显著,其组成的合金电极具有更高的电化学容量保持率. ...
Electrochemical characteristics of amorphous Mg0.9M0.1Ni (M = Ni, Ti, Zr, Co and Si) ternary alloys prepared by mechanical alloying
1
2000
... 合金化常被用来改善镁基非晶合金电极的循环性能.Ni[83]、V[84]、Co[85]、Ti[86]等作为非晶镁基合金化的典型代表元素,可以有效改善非晶合金的电化学循环性能.Anik等[86]制备的Mg0.9Ti0.2Ni0.9合金电极在20 cyc充放电循环后,容量保持率高达72%,表明金属Ti合金化对电极的循环稳定性提高显著.过去认为Ti的加入使得充/放电过程中合金表面形成的TiO2层延缓了合金电极的腐蚀速率.Han等[87]对Mg0.7Ti0.3Ni1.0合金的电化学阻抗分析表明,在充/放电循环过程中Mg0.7Ti0.3Ni1.0合金与电解液之间的电荷转移电阻不会增加,Auger电子能谱(AES)也表明Mg0.7Ti0.3Ni1.0合金电极表面氧化层的厚度要小于MgNi合金电极表面的氧化层厚度.事实上,Ti的主要作用是提高了非晶的稳定性,避免了氢致非晶化.Huang等[88]在非晶Mg0.50Ni0.50合金中引入Ti制备了Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金,使晶化温度由Mg0.50Ni0.50合金的300℃提高到350℃.由图7a[88]可以看出,Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后TG曲线的失重率分别为17.3%、18.7%和17.0%,相近的失重率说明腐蚀速率基本相同,也说明Ti添加并未有效改善合金电极的耐腐蚀性,这意味着添加Ti对循环性能的改善另有原因.图7b[88]为球磨60 h制备的Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后的DSC曲线,Mg0.50Ni0.50的2个峰分别对应脱氢峰和Mg(OH)2分解的吸热峰,而添加了金属Ti的电极没有脱氢峰的出现,说明添加Ti可以使合金在充/放电的过程中保持可逆的吸/放氢反应.而且在370℃左右处Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50电极合金均出现了非晶相的晶化转变峰,也说明了添加了Ti的合金经30 cyc循环后非晶相依旧得到了保留,而Mg0.50Ni0.50电极合金则已经完全晶化.上述结果很好地说明了添加Ti可以抑制合金非晶相的氢致晶化转变,从而提高Mg-Ni非晶合金电极的稳定性.值得注意的是,添加Ti还导致了合金中形成TiNi催化相,从而提高了非晶合金电极的倍率性能和动力学性能,但随着Ti添加量的增加,TiNi相增多而非晶相减少,导致合金的放电容量降低.随后,Li等[89]通过球磨法探讨了添加In、Al、Cu和Sc等对Mg-Ni基非晶合金电化学循环稳定性和热力学稳定性的影响,发现添加Al、Cu和In可以抑制合金电极在充/放电循环中的氢致晶化,其中添加Al的作用最显著,其组成的合金电极具有更高的电化学容量保持率. ...
Effect of Al, B, Ti and Zr additive elements on the electrochemical hydrogen storage performance of MgNi alloy
2
2011
... 合金化常被用来改善镁基非晶合金电极的循环性能.Ni[83]、V[84]、Co[85]、Ti[86]等作为非晶镁基合金化的典型代表元素,可以有效改善非晶合金的电化学循环性能.Anik等[86]制备的Mg0.9Ti0.2Ni0.9合金电极在20 cyc充放电循环后,容量保持率高达72%,表明金属Ti合金化对电极的循环稳定性提高显著.过去认为Ti的加入使得充/放电过程中合金表面形成的TiO2层延缓了合金电极的腐蚀速率.Han等[87]对Mg0.7Ti0.3Ni1.0合金的电化学阻抗分析表明,在充/放电循环过程中Mg0.7Ti0.3Ni1.0合金与电解液之间的电荷转移电阻不会增加,Auger电子能谱(AES)也表明Mg0.7Ti0.3Ni1.0合金电极表面氧化层的厚度要小于MgNi合金电极表面的氧化层厚度.事实上,Ti的主要作用是提高了非晶的稳定性,避免了氢致非晶化.Huang等[88]在非晶Mg0.50Ni0.50合金中引入Ti制备了Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金,使晶化温度由Mg0.50Ni0.50合金的300℃提高到350℃.由图7a[88]可以看出,Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后TG曲线的失重率分别为17.3%、18.7%和17.0%,相近的失重率说明腐蚀速率基本相同,也说明Ti添加并未有效改善合金电极的耐腐蚀性,这意味着添加Ti对循环性能的改善另有原因.图7b[88]为球磨60 h制备的Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后的DSC曲线,Mg0.50Ni0.50的2个峰分别对应脱氢峰和Mg(OH)2分解的吸热峰,而添加了金属Ti的电极没有脱氢峰的出现,说明添加Ti可以使合金在充/放电的过程中保持可逆的吸/放氢反应.而且在370℃左右处Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50电极合金均出现了非晶相的晶化转变峰,也说明了添加了Ti的合金经30 cyc循环后非晶相依旧得到了保留,而Mg0.50Ni0.50电极合金则已经完全晶化.上述结果很好地说明了添加Ti可以抑制合金非晶相的氢致晶化转变,从而提高Mg-Ni非晶合金电极的稳定性.值得注意的是,添加Ti还导致了合金中形成TiNi催化相,从而提高了非晶合金电极的倍率性能和动力学性能,但随着Ti添加量的增加,TiNi相增多而非晶相减少,导致合金的放电容量降低.随后,Li等[89]通过球磨法探讨了添加In、Al、Cu和Sc等对Mg-Ni基非晶合金电化学循环稳定性和热力学稳定性的影响,发现添加Al、Cu和In可以抑制合金电极在充/放电循环中的氢致晶化,其中添加Al的作用最显著,其组成的合金电极具有更高的电化学容量保持率. ...
... [86]制备的Mg0.9Ti0.2Ni0.9合金电极在20 cyc充放电循环后,容量保持率高达72%,表明金属Ti合金化对电极的循环稳定性提高显著.过去认为Ti的加入使得充/放电过程中合金表面形成的TiO2层延缓了合金电极的腐蚀速率.Han等[87]对Mg0.7Ti0.3Ni1.0合金的电化学阻抗分析表明,在充/放电循环过程中Mg0.7Ti0.3Ni1.0合金与电解液之间的电荷转移电阻不会增加,Auger电子能谱(AES)也表明Mg0.7Ti0.3Ni1.0合金电极表面氧化层的厚度要小于MgNi合金电极表面的氧化层厚度.事实上,Ti的主要作用是提高了非晶的稳定性,避免了氢致非晶化.Huang等[88]在非晶Mg0.50Ni0.50合金中引入Ti制备了Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金,使晶化温度由Mg0.50Ni0.50合金的300℃提高到350℃.由图7a[88]可以看出,Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后TG曲线的失重率分别为17.3%、18.7%和17.0%,相近的失重率说明腐蚀速率基本相同,也说明Ti添加并未有效改善合金电极的耐腐蚀性,这意味着添加Ti对循环性能的改善另有原因.图7b[88]为球磨60 h制备的Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后的DSC曲线,Mg0.50Ni0.50的2个峰分别对应脱氢峰和Mg(OH)2分解的吸热峰,而添加了金属Ti的电极没有脱氢峰的出现,说明添加Ti可以使合金在充/放电的过程中保持可逆的吸/放氢反应.而且在370℃左右处Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50电极合金均出现了非晶相的晶化转变峰,也说明了添加了Ti的合金经30 cyc循环后非晶相依旧得到了保留,而Mg0.50Ni0.50电极合金则已经完全晶化.上述结果很好地说明了添加Ti可以抑制合金非晶相的氢致晶化转变,从而提高Mg-Ni非晶合金电极的稳定性.值得注意的是,添加Ti还导致了合金中形成TiNi催化相,从而提高了非晶合金电极的倍率性能和动力学性能,但随着Ti添加量的增加,TiNi相增多而非晶相减少,导致合金的放电容量降低.随后,Li等[89]通过球磨法探讨了添加In、Al、Cu和Sc等对Mg-Ni基非晶合金电化学循环稳定性和热力学稳定性的影响,发现添加Al、Cu和In可以抑制合金电极在充/放电循环中的氢致晶化,其中添加Al的作用最显著,其组成的合金电极具有更高的电化学容量保持率. ...
Effects of Ti on the cycle life of amorphous MgNi-based alloy prepared by ball milling
1
2000
... 合金化常被用来改善镁基非晶合金电极的循环性能.Ni[83]、V[84]、Co[85]、Ti[86]等作为非晶镁基合金化的典型代表元素,可以有效改善非晶合金的电化学循环性能.Anik等[86]制备的Mg0.9Ti0.2Ni0.9合金电极在20 cyc充放电循环后,容量保持率高达72%,表明金属Ti合金化对电极的循环稳定性提高显著.过去认为Ti的加入使得充/放电过程中合金表面形成的TiO2层延缓了合金电极的腐蚀速率.Han等[87]对Mg0.7Ti0.3Ni1.0合金的电化学阻抗分析表明,在充/放电循环过程中Mg0.7Ti0.3Ni1.0合金与电解液之间的电荷转移电阻不会增加,Auger电子能谱(AES)也表明Mg0.7Ti0.3Ni1.0合金电极表面氧化层的厚度要小于MgNi合金电极表面的氧化层厚度.事实上,Ti的主要作用是提高了非晶的稳定性,避免了氢致非晶化.Huang等[88]在非晶Mg0.50Ni0.50合金中引入Ti制备了Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金,使晶化温度由Mg0.50Ni0.50合金的300℃提高到350℃.由图7a[88]可以看出,Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后TG曲线的失重率分别为17.3%、18.7%和17.0%,相近的失重率说明腐蚀速率基本相同,也说明Ti添加并未有效改善合金电极的耐腐蚀性,这意味着添加Ti对循环性能的改善另有原因.图7b[88]为球磨60 h制备的Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后的DSC曲线,Mg0.50Ni0.50的2个峰分别对应脱氢峰和Mg(OH)2分解的吸热峰,而添加了金属Ti的电极没有脱氢峰的出现,说明添加Ti可以使合金在充/放电的过程中保持可逆的吸/放氢反应.而且在370℃左右处Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50电极合金均出现了非晶相的晶化转变峰,也说明了添加了Ti的合金经30 cyc循环后非晶相依旧得到了保留,而Mg0.50Ni0.50电极合金则已经完全晶化.上述结果很好地说明了添加Ti可以抑制合金非晶相的氢致晶化转变,从而提高Mg-Ni非晶合金电极的稳定性.值得注意的是,添加Ti还导致了合金中形成TiNi催化相,从而提高了非晶合金电极的倍率性能和动力学性能,但随着Ti添加量的增加,TiNi相增多而非晶相减少,导致合金的放电容量降低.随后,Li等[89]通过球磨法探讨了添加In、Al、Cu和Sc等对Mg-Ni基非晶合金电化学循环稳定性和热力学稳定性的影响,发现添加Al、Cu和In可以抑制合金电极在充/放电循环中的氢致晶化,其中添加Al的作用最显著,其组成的合金电极具有更高的电化学容量保持率. ...
Reducing the electrochemical capacity decay of milled Mg-Ni alloys: The role of stabilizing amorphous phase by Ti-substitution
5
2019
... 合金化常被用来改善镁基非晶合金电极的循环性能.Ni[83]、V[84]、Co[85]、Ti[86]等作为非晶镁基合金化的典型代表元素,可以有效改善非晶合金的电化学循环性能.Anik等[86]制备的Mg0.9Ti0.2Ni0.9合金电极在20 cyc充放电循环后,容量保持率高达72%,表明金属Ti合金化对电极的循环稳定性提高显著.过去认为Ti的加入使得充/放电过程中合金表面形成的TiO2层延缓了合金电极的腐蚀速率.Han等[87]对Mg0.7Ti0.3Ni1.0合金的电化学阻抗分析表明,在充/放电循环过程中Mg0.7Ti0.3Ni1.0合金与电解液之间的电荷转移电阻不会增加,Auger电子能谱(AES)也表明Mg0.7Ti0.3Ni1.0合金电极表面氧化层的厚度要小于MgNi合金电极表面的氧化层厚度.事实上,Ti的主要作用是提高了非晶的稳定性,避免了氢致非晶化.Huang等[88]在非晶Mg0.50Ni0.50合金中引入Ti制备了Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金,使晶化温度由Mg0.50Ni0.50合金的300℃提高到350℃.由图7a[88]可以看出,Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后TG曲线的失重率分别为17.3%、18.7%和17.0%,相近的失重率说明腐蚀速率基本相同,也说明Ti添加并未有效改善合金电极的耐腐蚀性,这意味着添加Ti对循环性能的改善另有原因.图7b[88]为球磨60 h制备的Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后的DSC曲线,Mg0.50Ni0.50的2个峰分别对应脱氢峰和Mg(OH)2分解的吸热峰,而添加了金属Ti的电极没有脱氢峰的出现,说明添加Ti可以使合金在充/放电的过程中保持可逆的吸/放氢反应.而且在370℃左右处Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50电极合金均出现了非晶相的晶化转变峰,也说明了添加了Ti的合金经30 cyc循环后非晶相依旧得到了保留,而Mg0.50Ni0.50电极合金则已经完全晶化.上述结果很好地说明了添加Ti可以抑制合金非晶相的氢致晶化转变,从而提高Mg-Ni非晶合金电极的稳定性.值得注意的是,添加Ti还导致了合金中形成TiNi催化相,从而提高了非晶合金电极的倍率性能和动力学性能,但随着Ti添加量的增加,TiNi相增多而非晶相减少,导致合金的放电容量降低.随后,Li等[89]通过球磨法探讨了添加In、Al、Cu和Sc等对Mg-Ni基非晶合金电化学循环稳定性和热力学稳定性的影响,发现添加Al、Cu和In可以抑制合金电极在充/放电循环中的氢致晶化,其中添加Al的作用最显著,其组成的合金电极具有更高的电化学容量保持率. ...
... [88]可以看出,Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后TG曲线的失重率分别为17.3%、18.7%和17.0%,相近的失重率说明腐蚀速率基本相同,也说明Ti添加并未有效改善合金电极的耐腐蚀性,这意味着添加Ti对循环性能的改善另有原因.图7b[88]为球磨60 h制备的Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后的DSC曲线,Mg0.50Ni0.50的2个峰分别对应脱氢峰和Mg(OH)2分解的吸热峰,而添加了金属Ti的电极没有脱氢峰的出现,说明添加Ti可以使合金在充/放电的过程中保持可逆的吸/放氢反应.而且在370℃左右处Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50电极合金均出现了非晶相的晶化转变峰,也说明了添加了Ti的合金经30 cyc循环后非晶相依旧得到了保留,而Mg0.50Ni0.50电极合金则已经完全晶化.上述结果很好地说明了添加Ti可以抑制合金非晶相的氢致晶化转变,从而提高Mg-Ni非晶合金电极的稳定性.值得注意的是,添加Ti还导致了合金中形成TiNi催化相,从而提高了非晶合金电极的倍率性能和动力学性能,但随着Ti添加量的增加,TiNi相增多而非晶相减少,导致合金的放电容量降低.随后,Li等[89]通过球磨法探讨了添加In、Al、Cu和Sc等对Mg-Ni基非晶合金电化学循环稳定性和热力学稳定性的影响,发现添加Al、Cu和In可以抑制合金电极在充/放电循环中的氢致晶化,其中添加Al的作用最显著,其组成的合金电极具有更高的电化学容量保持率. ...
... [88]为球磨60 h制备的Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后的DSC曲线,Mg0.50Ni0.50的2个峰分别对应脱氢峰和Mg(OH)2分解的吸热峰,而添加了金属Ti的电极没有脱氢峰的出现,说明添加Ti可以使合金在充/放电的过程中保持可逆的吸/放氢反应.而且在370℃左右处Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50电极合金均出现了非晶相的晶化转变峰,也说明了添加了Ti的合金经30 cyc循环后非晶相依旧得到了保留,而Mg0.50Ni0.50电极合金则已经完全晶化.上述结果很好地说明了添加Ti可以抑制合金非晶相的氢致晶化转变,从而提高Mg-Ni非晶合金电极的稳定性.值得注意的是,添加Ti还导致了合金中形成TiNi催化相,从而提高了非晶合金电极的倍率性能和动力学性能,但随着Ti添加量的增加,TiNi相增多而非晶相减少,导致合金的放电容量降低.随后,Li等[89]通过球磨法探讨了添加In、Al、Cu和Sc等对Mg-Ni基非晶合金电化学循环稳定性和热力学稳定性的影响,发现添加Al、Cu和In可以抑制合金电极在充/放电循环中的氢致晶化,其中添加Al的作用最显著,其组成的合金电极具有更高的电化学容量保持率. ...
... [
88]
TG (a) and DSC (b) results of the 60 h milled Mg<sub>0.50</sub>Ni<sub>0.50</sub>, Mg<sub>0.45</sub>Ti<sub>0.05</sub>Ni<sub>0.50</sub>, and Mg<sub>0.40</sub>Ti<sub>0.10</sub>Ni<sub>0.50</sub> alloy electrodes at discharge state after 30 cyc<sup>[<xref ref-type="bibr" rid="R88">88</xref>]</sup>Fig.7![]()
解决氢致晶化带来的衰减仍不能避免Mg-Ni非晶合金电极的衰减问题,镁基非晶合金在充/放电循环中的腐蚀依然十分严重.为克服腐蚀的问题,开发针对于镁基非晶合金的新型电解液亦不失为一种有效策略.在传统6 mol/L KOH电解液基础上添加少量盐溶液是最常见的改性手段[90~93].Kim等[90]在碱性电解液中添加少量KF,观察到Mg2Ni合金电极表面在充放电过程中会形成可以有效抑制Mg(OH)2生成的活性MgF2层,使得合金电极的循环寿命有所提高.Mohamad等[94]将聚合物引入电解液中,以添加60%PVA的PVA-KOH聚合物电解质为电解液,采用机械合金化制备的Mg2Ni合金,在0.1 mA的恒定电流放电时的放电电压可以在1.2 V以上的平台电压保持约10 h,降低了自放电速率.毋庸置疑,基于KOH电解液的改性很难从源头上避免KOH溶液对腐蚀电位较低的金属Mg的腐蚀,探索对金属Mg有较低腐蚀性的新型电解液十分必要.Huang等[95]探索了采用四甲基氢氧化铵(TMAH)溶液作为电解液的效果.相较于6 mol/L KOH溶液,接近饱和浓度的4.5 mol/L TMAH溶液对于Mg0.4Ti0.1Ni0.5合金电极的腐蚀性更小,并且可以满足Mg0.4Ti0.1Ni0.5负极和Ni(OH)2正极发生氧化还原反应的条件,从而使Mg0.4Ti0.1Ni0.5合金电极在TMAH溶液中表现出更好的循环稳定性.更进一步,在4.5 mol/L TMAH电解液中加入少量Cu(OH)2 (4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2),使得Cu在前几次充/放电循环中原位沉积在合金电极表面,Mg0.4Ti0.1Ni0.5合金电极在4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2溶液中经过100 cyc充/放电循环后,放电容量依然保持在313 mAh/g,循环寿命提高十分显著[95]. ...
... [
88]
Fig.7![]()
解决氢致晶化带来的衰减仍不能避免Mg-Ni非晶合金电极的衰减问题,镁基非晶合金在充/放电循环中的腐蚀依然十分严重.为克服腐蚀的问题,开发针对于镁基非晶合金的新型电解液亦不失为一种有效策略.在传统6 mol/L KOH电解液基础上添加少量盐溶液是最常见的改性手段[90~93].Kim等[90]在碱性电解液中添加少量KF,观察到Mg2Ni合金电极表面在充放电过程中会形成可以有效抑制Mg(OH)2生成的活性MgF2层,使得合金电极的循环寿命有所提高.Mohamad等[94]将聚合物引入电解液中,以添加60%PVA的PVA-KOH聚合物电解质为电解液,采用机械合金化制备的Mg2Ni合金,在0.1 mA的恒定电流放电时的放电电压可以在1.2 V以上的平台电压保持约10 h,降低了自放电速率.毋庸置疑,基于KOH电解液的改性很难从源头上避免KOH溶液对腐蚀电位较低的金属Mg的腐蚀,探索对金属Mg有较低腐蚀性的新型电解液十分必要.Huang等[95]探索了采用四甲基氢氧化铵(TMAH)溶液作为电解液的效果.相较于6 mol/L KOH溶液,接近饱和浓度的4.5 mol/L TMAH溶液对于Mg0.4Ti0.1Ni0.5合金电极的腐蚀性更小,并且可以满足Mg0.4Ti0.1Ni0.5负极和Ni(OH)2正极发生氧化还原反应的条件,从而使Mg0.4Ti0.1Ni0.5合金电极在TMAH溶液中表现出更好的循环稳定性.更进一步,在4.5 mol/L TMAH电解液中加入少量Cu(OH)2 (4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2),使得Cu在前几次充/放电循环中原位沉积在合金电极表面,Mg0.4Ti0.1Ni0.5合金电极在4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2溶液中经过100 cyc充/放电循环后,放电容量依然保持在313 mAh/g,循环寿命提高十分显著[95]. ...
Promoting the cycling stability of amorphous MgNi-based alloy electrodes by mitigating hydrogen-induced crystallization
1
2021
... 合金化常被用来改善镁基非晶合金电极的循环性能.Ni[83]、V[84]、Co[85]、Ti[86]等作为非晶镁基合金化的典型代表元素,可以有效改善非晶合金的电化学循环性能.Anik等[86]制备的Mg0.9Ti0.2Ni0.9合金电极在20 cyc充放电循环后,容量保持率高达72%,表明金属Ti合金化对电极的循环稳定性提高显著.过去认为Ti的加入使得充/放电过程中合金表面形成的TiO2层延缓了合金电极的腐蚀速率.Han等[87]对Mg0.7Ti0.3Ni1.0合金的电化学阻抗分析表明,在充/放电循环过程中Mg0.7Ti0.3Ni1.0合金与电解液之间的电荷转移电阻不会增加,Auger电子能谱(AES)也表明Mg0.7Ti0.3Ni1.0合金电极表面氧化层的厚度要小于MgNi合金电极表面的氧化层厚度.事实上,Ti的主要作用是提高了非晶的稳定性,避免了氢致非晶化.Huang等[88]在非晶Mg0.50Ni0.50合金中引入Ti制备了Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金,使晶化温度由Mg0.50Ni0.50合金的300℃提高到350℃.由图7a[88]可以看出,Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后TG曲线的失重率分别为17.3%、18.7%和17.0%,相近的失重率说明腐蚀速率基本相同,也说明Ti添加并未有效改善合金电极的耐腐蚀性,这意味着添加Ti对循环性能的改善另有原因.图7b[88]为球磨60 h制备的Mg0.50Ni0.50、Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50合金电极经30 cyc循环后的DSC曲线,Mg0.50Ni0.50的2个峰分别对应脱氢峰和Mg(OH)2分解的吸热峰,而添加了金属Ti的电极没有脱氢峰的出现,说明添加Ti可以使合金在充/放电的过程中保持可逆的吸/放氢反应.而且在370℃左右处Mg0.45Ti0.05Ni0.50和Mg0.40Ti0.10Ni0.50电极合金均出现了非晶相的晶化转变峰,也说明了添加了Ti的合金经30 cyc循环后非晶相依旧得到了保留,而Mg0.50Ni0.50电极合金则已经完全晶化.上述结果很好地说明了添加Ti可以抑制合金非晶相的氢致晶化转变,从而提高Mg-Ni非晶合金电极的稳定性.值得注意的是,添加Ti还导致了合金中形成TiNi催化相,从而提高了非晶合金电极的倍率性能和动力学性能,但随着Ti添加量的增加,TiNi相增多而非晶相减少,导致合金的放电容量降低.随后,Li等[89]通过球磨法探讨了添加In、Al、Cu和Sc等对Mg-Ni基非晶合金电化学循环稳定性和热力学稳定性的影响,发现添加Al、Cu和In可以抑制合金电极在充/放电循环中的氢致晶化,其中添加Al的作用最显著,其组成的合金电极具有更高的电化学容量保持率. ...
Effects of F-treatment on degradation of Mg2Ni electrode fabricated by mechanical alloying
2
2002
... 解决氢致晶化带来的衰减仍不能避免Mg-Ni非晶合金电极的衰减问题,镁基非晶合金在充/放电循环中的腐蚀依然十分严重.为克服腐蚀的问题,开发针对于镁基非晶合金的新型电解液亦不失为一种有效策略.在传统6 mol/L KOH电解液基础上添加少量盐溶液是最常见的改性手段[90~93].Kim等[90]在碱性电解液中添加少量KF,观察到Mg2Ni合金电极表面在充放电过程中会形成可以有效抑制Mg(OH)2生成的活性MgF2层,使得合金电极的循环寿命有所提高.Mohamad等[94]将聚合物引入电解液中,以添加60%PVA的PVA-KOH聚合物电解质为电解液,采用机械合金化制备的Mg2Ni合金,在0.1 mA的恒定电流放电时的放电电压可以在1.2 V以上的平台电压保持约10 h,降低了自放电速率.毋庸置疑,基于KOH电解液的改性很难从源头上避免KOH溶液对腐蚀电位较低的金属Mg的腐蚀,探索对金属Mg有较低腐蚀性的新型电解液十分必要.Huang等[95]探索了采用四甲基氢氧化铵(TMAH)溶液作为电解液的效果.相较于6 mol/L KOH溶液,接近饱和浓度的4.5 mol/L TMAH溶液对于Mg0.4Ti0.1Ni0.5合金电极的腐蚀性更小,并且可以满足Mg0.4Ti0.1Ni0.5负极和Ni(OH)2正极发生氧化还原反应的条件,从而使Mg0.4Ti0.1Ni0.5合金电极在TMAH溶液中表现出更好的循环稳定性.更进一步,在4.5 mol/L TMAH电解液中加入少量Cu(OH)2 (4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2),使得Cu在前几次充/放电循环中原位沉积在合金电极表面,Mg0.4Ti0.1Ni0.5合金电极在4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2溶液中经过100 cyc充/放电循环后,放电容量依然保持在313 mAh/g,循环寿命提高十分显著[95]. ...
... [90]在碱性电解液中添加少量KF,观察到Mg2Ni合金电极表面在充放电过程中会形成可以有效抑制Mg(OH)2生成的活性MgF2层,使得合金电极的循环寿命有所提高.Mohamad等[94]将聚合物引入电解液中,以添加60%PVA的PVA-KOH聚合物电解质为电解液,采用机械合金化制备的Mg2Ni合金,在0.1 mA的恒定电流放电时的放电电压可以在1.2 V以上的平台电压保持约10 h,降低了自放电速率.毋庸置疑,基于KOH电解液的改性很难从源头上避免KOH溶液对腐蚀电位较低的金属Mg的腐蚀,探索对金属Mg有较低腐蚀性的新型电解液十分必要.Huang等[95]探索了采用四甲基氢氧化铵(TMAH)溶液作为电解液的效果.相较于6 mol/L KOH溶液,接近饱和浓度的4.5 mol/L TMAH溶液对于Mg0.4Ti0.1Ni0.5合金电极的腐蚀性更小,并且可以满足Mg0.4Ti0.1Ni0.5负极和Ni(OH)2正极发生氧化还原反应的条件,从而使Mg0.4Ti0.1Ni0.5合金电极在TMAH溶液中表现出更好的循环稳定性.更进一步,在4.5 mol/L TMAH电解液中加入少量Cu(OH)2 (4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2),使得Cu在前几次充/放电循环中原位沉积在合金电极表面,Mg0.4Ti0.1Ni0.5合金电极在4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2溶液中经过100 cyc充/放电循环后,放电容量依然保持在313 mAh/g,循环寿命提高十分显著[95]. ...
Effects of Cs2CO3 additive in KOH electrolyte used in Ni/MH batteries
0
2017
Effects of salt additives to the KOH electrolyte used in Ni/MH batteries
0
2015
Sodium tungstate as electrolyte additive to improve high-temperature performance of nickel-metal hydride batteries
1
2013
... 解决氢致晶化带来的衰减仍不能避免Mg-Ni非晶合金电极的衰减问题,镁基非晶合金在充/放电循环中的腐蚀依然十分严重.为克服腐蚀的问题,开发针对于镁基非晶合金的新型电解液亦不失为一种有效策略.在传统6 mol/L KOH电解液基础上添加少量盐溶液是最常见的改性手段[90~93].Kim等[90]在碱性电解液中添加少量KF,观察到Mg2Ni合金电极表面在充放电过程中会形成可以有效抑制Mg(OH)2生成的活性MgF2层,使得合金电极的循环寿命有所提高.Mohamad等[94]将聚合物引入电解液中,以添加60%PVA的PVA-KOH聚合物电解质为电解液,采用机械合金化制备的Mg2Ni合金,在0.1 mA的恒定电流放电时的放电电压可以在1.2 V以上的平台电压保持约10 h,降低了自放电速率.毋庸置疑,基于KOH电解液的改性很难从源头上避免KOH溶液对腐蚀电位较低的金属Mg的腐蚀,探索对金属Mg有较低腐蚀性的新型电解液十分必要.Huang等[95]探索了采用四甲基氢氧化铵(TMAH)溶液作为电解液的效果.相较于6 mol/L KOH溶液,接近饱和浓度的4.5 mol/L TMAH溶液对于Mg0.4Ti0.1Ni0.5合金电极的腐蚀性更小,并且可以满足Mg0.4Ti0.1Ni0.5负极和Ni(OH)2正极发生氧化还原反应的条件,从而使Mg0.4Ti0.1Ni0.5合金电极在TMAH溶液中表现出更好的循环稳定性.更进一步,在4.5 mol/L TMAH电解液中加入少量Cu(OH)2 (4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2),使得Cu在前几次充/放电循环中原位沉积在合金电极表面,Mg0.4Ti0.1Ni0.5合金电极在4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2溶液中经过100 cyc充/放电循环后,放电容量依然保持在313 mAh/g,循环寿命提高十分显著[95]. ...
Studies of alkaline solid polymer electrolyte and mechanically alloyed polycrystalline Mg2Ni for use in nickel metal hydride batteries
1
2002
... 解决氢致晶化带来的衰减仍不能避免Mg-Ni非晶合金电极的衰减问题,镁基非晶合金在充/放电循环中的腐蚀依然十分严重.为克服腐蚀的问题,开发针对于镁基非晶合金的新型电解液亦不失为一种有效策略.在传统6 mol/L KOH电解液基础上添加少量盐溶液是最常见的改性手段[90~93].Kim等[90]在碱性电解液中添加少量KF,观察到Mg2Ni合金电极表面在充放电过程中会形成可以有效抑制Mg(OH)2生成的活性MgF2层,使得合金电极的循环寿命有所提高.Mohamad等[94]将聚合物引入电解液中,以添加60%PVA的PVA-KOH聚合物电解质为电解液,采用机械合金化制备的Mg2Ni合金,在0.1 mA的恒定电流放电时的放电电压可以在1.2 V以上的平台电压保持约10 h,降低了自放电速率.毋庸置疑,基于KOH电解液的改性很难从源头上避免KOH溶液对腐蚀电位较低的金属Mg的腐蚀,探索对金属Mg有较低腐蚀性的新型电解液十分必要.Huang等[95]探索了采用四甲基氢氧化铵(TMAH)溶液作为电解液的效果.相较于6 mol/L KOH溶液,接近饱和浓度的4.5 mol/L TMAH溶液对于Mg0.4Ti0.1Ni0.5合金电极的腐蚀性更小,并且可以满足Mg0.4Ti0.1Ni0.5负极和Ni(OH)2正极发生氧化还原反应的条件,从而使Mg0.4Ti0.1Ni0.5合金电极在TMAH溶液中表现出更好的循环稳定性.更进一步,在4.5 mol/L TMAH电解液中加入少量Cu(OH)2 (4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2),使得Cu在前几次充/放电循环中原位沉积在合金电极表面,Mg0.4Ti0.1Ni0.5合金电极在4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2溶液中经过100 cyc充/放电循环后,放电容量依然保持在313 mAh/g,循环寿命提高十分显著[95]. ...
Exploring new electrolyte to inhibit corrosion of Mg-based amorphous alloy anodes: A route for promotion energy density of Ni-MH battery
2
... 解决氢致晶化带来的衰减仍不能避免Mg-Ni非晶合金电极的衰减问题,镁基非晶合金在充/放电循环中的腐蚀依然十分严重.为克服腐蚀的问题,开发针对于镁基非晶合金的新型电解液亦不失为一种有效策略.在传统6 mol/L KOH电解液基础上添加少量盐溶液是最常见的改性手段[90~93].Kim等[90]在碱性电解液中添加少量KF,观察到Mg2Ni合金电极表面在充放电过程中会形成可以有效抑制Mg(OH)2生成的活性MgF2层,使得合金电极的循环寿命有所提高.Mohamad等[94]将聚合物引入电解液中,以添加60%PVA的PVA-KOH聚合物电解质为电解液,采用机械合金化制备的Mg2Ni合金,在0.1 mA的恒定电流放电时的放电电压可以在1.2 V以上的平台电压保持约10 h,降低了自放电速率.毋庸置疑,基于KOH电解液的改性很难从源头上避免KOH溶液对腐蚀电位较低的金属Mg的腐蚀,探索对金属Mg有较低腐蚀性的新型电解液十分必要.Huang等[95]探索了采用四甲基氢氧化铵(TMAH)溶液作为电解液的效果.相较于6 mol/L KOH溶液,接近饱和浓度的4.5 mol/L TMAH溶液对于Mg0.4Ti0.1Ni0.5合金电极的腐蚀性更小,并且可以满足Mg0.4Ti0.1Ni0.5负极和Ni(OH)2正极发生氧化还原反应的条件,从而使Mg0.4Ti0.1Ni0.5合金电极在TMAH溶液中表现出更好的循环稳定性.更进一步,在4.5 mol/L TMAH电解液中加入少量Cu(OH)2 (4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2),使得Cu在前几次充/放电循环中原位沉积在合金电极表面,Mg0.4Ti0.1Ni0.5合金电极在4.5 mol/L TMAH + 0.01 mol/L Cu(OH)2溶液中经过100 cyc充/放电循环后,放电容量依然保持在313 mAh/g,循环寿命提高十分显著[95]. ...
... [95]. ...



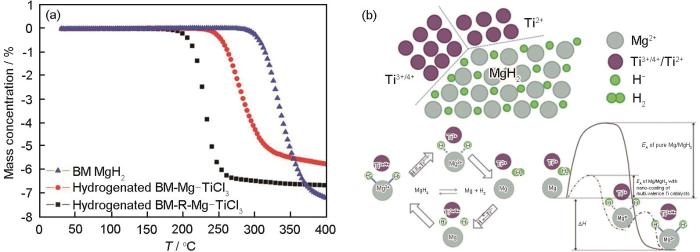


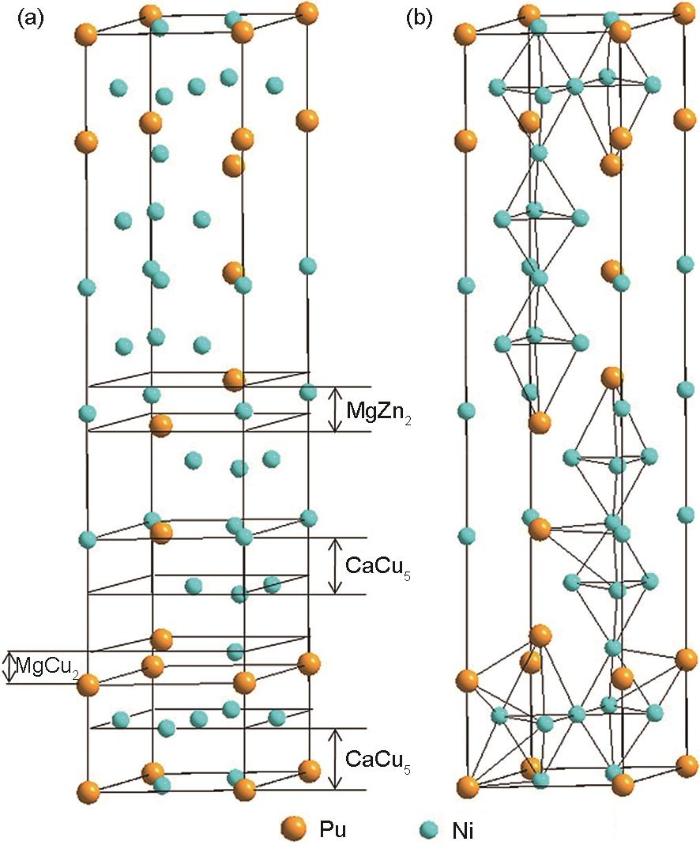
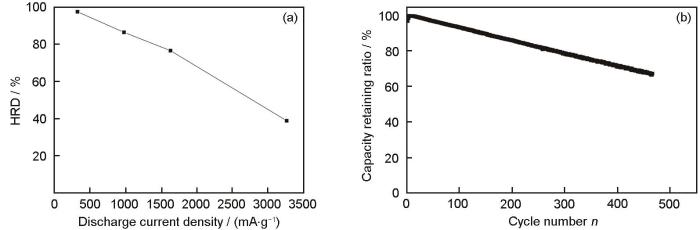
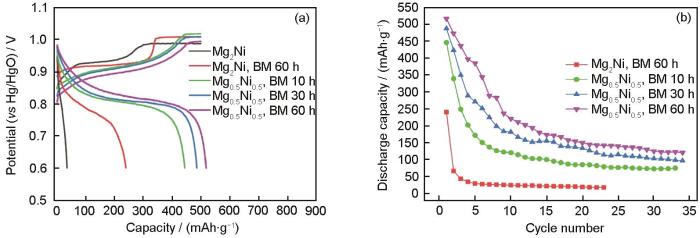
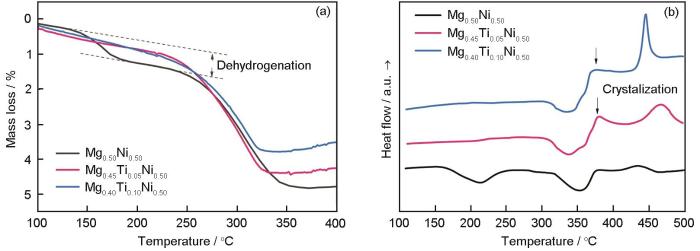

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}