


从最初依赖经验、观察和实验研究,到热力学定律等以数学建模为特征的理论研究,到结合材料理论、物理、化学等交叉学科的计算模拟研究,再到以材料高通量实验、计算和表征以及数据库为基础的材料基因工程,每一次进展都拓展了我们对材料学领域的认知边界。如今,蓬勃发展的人工智能方法已然成为推动材料研究的新范式[1],旨在通过机器学习等人工智能手段,深度挖掘材料成分-工艺-组织-性能之间的复杂关联,显著加速传统材料的改良,以及新材料、新性能的发现和设计。
在材料的计算模拟研究中,原子尺度的计算模拟是连接微观结构与宏观性能的关键桥梁[2~4]。然而,该领域长期面临着核心挑战:计算精度与效率的“两难困境”。一方面,基于第一性原理的计算方法虽然精度高,但其高昂的计算成本将其应用限制在数百个原子和皮秒级的时间尺度内;另一方面,诸如嵌入原子势(embedded atom method,EAM)[5,6]、修正嵌入原子势(modified embedded atom method,MEAM)[7]、Tersoff[8]等经验力场,虽然能够实现百万甚至上亿原子体系的纳秒级模拟,但其往往采用基于物理近似的简化模型,导致精度和适用性存在固有局限。此外,这类力场的构建过程依赖对体系物理特性的先验认知,难以实现普适性应用。
为解决上述瓶颈问题,研究者将数据和智能驱动新范式思想与传统的计算模拟深度融合,催生了机器学习势(machine learning potential,MLP)函数,又称作机器学习力场(machine-learned force field,MLFF)。其核心思想不再是依赖物理直觉去构建固定的函数形式,而是借助灵活的机器学习模型(如神经网络),直接从海量的第一性原理计算数据中“学习”高维、高精度的势能面(potential energy surface,PES)。这种数据驱动的方法为原子间势函数的开发带来了革命性突破,有效解决了计算模拟中长期存在的精度与效率不可兼得的难题。
回顾MLFF的发展历程,其早期的探索可追溯到20世纪90年代,研究人员[9~14]尝试使用神经网络拟合小分子体系的势能面。然而,这些早期模型通常采用体系的整体坐标作为输入,导致模型仅限于特定的小体系,缺乏处理原子置换不变性的内在机制和向大体系迁移的能力。2007年,Behler和Parrinello[15]在其开创性的工作中提出了一种高维神经网络势(high dimensional neural network potential,HDNNP),标志着MLFF发展的范式转变。他们提出的两大核心思想——“局域性假设”(将体系总能量分解为各原子能量贡献之和)和原子中心对称函数(atom-centered symmetry function,ACSF)描述符,成功解决了早期方法的根本局限。ACSF将每个原子的局部化学环境编码为满足平移、旋转及同类原子置换不变性的固定维度特征向量,从而攻克了维度灾难和迁移性难题,使MLFF从理论探索正式迈入大规模实际应用阶段。MLFF的主要构建流程见图1,包括构建训练结构集合,对训练集进行第一性原理计算以得到其能量、力、应力张量等信息,选择合适的回归模型进行拟合,以及验证模型的准确性及泛化性能。
图1

图1
机器学习力场构建流程
Fig.1
Workflow of machine-learned force field (MLFF) construction (HEA—high-entropy alloy; F(x*)—atomic energy; x1-xn —reference local configurations; x*—local configuration to be predicted; ω1-ωN —model weights; K—kernel function; RC1 and RC2—coordinates; G1 and G2—local structure descriptors;
在Behler-Parrinello框架的启发下,MLFF的发展分化为两条主流技术路线。第一条路线致力于改进和发展更优的局域环境描述符和回归模型。在此路线中,基于核函数的方法取得了巨大成功,特别是Bartók等[16,17]发展的Gaussian近似势(Gaussian approximation potential,GAP),其采用的平滑原子位置重叠(smooth overlap of atomic positions,SOAP)描述符[18]已成为局域结构分析的最重要工具之一。Jinnouchi等[19,20]对该模型进行重要改进,通过在第一性原理分子动力学模拟中引入基于Bayesian力误差的实时主动学习机制,显著提升了训练集构建的智能化程度。除基于核函数的方法外,Shapeev[21]提出的基于线性回归的矩张量势(MTP)模型,Thompson等[22]提出的基于线性回归的SNAP (spectral neighbor analysis potential)模型,Wood和Thompson[23]提出的二次多项式回归的qSNAP (quaderatic spectral neighbor analysis potential)模型,以及Drautz[24]提出的原子团簇展开(atomic cluster expansion,ACE)模型,均以兼备高精度和高效率而获得了巨大成功。此外,刘智攀团队基于ACSF类型描述符开发了指数型描述符(power type structural descriptors,PTSDs),开发了LASP软件[25,26],在催化[27,28]、结构搜索[29,30]等领域获得了广泛应用。Behler团队[31~33]也进一步发展了他们构建的HDNNP模型,先后提出了四代模型,进一步引入电荷转移,整合了长程的Coulomb相互作用及磁矩等物理量。
第二条技术路线则寻求摆脱人为设计描述符的束缚,发展“端到端”的深度学习模型,让网络直接从原子坐标和种类中自动学习特征表示。深度势能(deep potential,DP)模型[34,35]是这一路线的杰出代表,它采用一个可训练的嵌入网络生成描述符,并在凝固、相变等诸多体系中取得了巨大成功[36~38]。图神经网络(graph neural network,GNN)模型则是该路线的另一类重要范式。其核心思想是将分子或材料结构表示为图,其中原子作为节点、化学键或邻近关系作为边,再通过迭代的信息传递和聚合机制更新节点特征,从而捕捉复杂的局域和非局域相互作用。典型的消息传递神经网络(message passing neural network,MPNN)框架[39]为后续工作奠定了基础,它通过在节点间交换“消息”,实现原子间相互作用的建模,并能够自然满足平移、旋转和置换等物理对称性。此方面开创性的工作为2018年Schütt等[40]提出的SchNet,并进一步发展出了PhysNet[41]、DimeNet (directional message passing neural network)[42]等一系列改进版本,这些方法通过设计更精细的消息函数或结合角度与三体信息进一步提升了准确性和泛化性。
近年来,MLFF的发展前沿聚焦于将物理知识嵌入模型架构。等变神经网络(equivariant neural network)为这一趋势的典型代表,其通过在网络结构中内置旋转等变性,显著提升了模型的数据利用效率和预测精度。这方面的代表性工作包括PaiNN[43]、NEQUIP[44]、Allegro[45]、MACE[46]、EquiRANN[47]、BOTNet[48]等,均展现出优于传统方法的性能。另一方面,针对长程相互作用建模的挑战,CACE-LES (cartesian atomic cluster expansion with latent Ewald summation)等模型[49]通过引入隐式Ewald求和技术,在不依赖显式电荷模型的前提下成功实现了对Coulomb、Van der Waals等长程相互作用的描述,并进一步拓展至材料极化性质的建模,为复杂材料体系的精确原子模拟提供了新的技术路径。
上述的机器学习势函数模型针对特定体系构建,开发和验证过程需要大量的人力和计算资源。针对该问题,机器学习势函数发展的另一个重要方向在于构建能够涵盖周期表元素的通用大模型(universal/foundation models)。此类模型旨在通过统一架构,实现对多种材料体系的普适性建模。借助Materials Project (MP)[50]等公开数据库中丰富的第一性原理数据,研究人员成功开发出诸如MEGNet[51]、M3GNet[52]、CHGNet[53]、GNoME[54]等一系列通用势函数模型。在DP模型基础上发展的DPA-1[55]和DPA-2[56]等模型进一步引入了注意力机制和预训练策略,显著增强了模型的泛化能力。基于MACE框架构建的MACE-MP-0模型[57],具备直接用于定性或半定量模拟的能力,也可作为基础模型,通过在特定任务数据上进行微调以达到更高的精度要求。此外,Xie等[58]开发的GPTFF (graph-based pre-trained transformer force field)模型将Transformer架构引入了模型中,进一步提升了模型的表达能力。典型的机器学习势函数模型发展过程见图2。
图2

图2
典型的机器学习势函数发展过程
Fig.2
Evolution of typical MLFF models (NN—neural network, MPNN—message passing neural network, KRR—kernel ridge regression, ETN—equivariant tensor network, HDNNP—high dimensional neural network potential, MSA—monomial symmetrization algorithm, GAP—Gaussian approximation potential, SNAP—spectral neighbor analysis method, MTP—moment tensor potential, Amp—atomistic machine-learning package, GDML—gradient domain machine learning, PROPhet—PROPerty prophet, DTNN—deep tensor neural network, qSNAP—quaderatic spectral neighbor analysis method, DP—deep potential, ACE—atomic cluster expansion, FCHL—Fabe-Christensen-Huang-Lilienfeld, LASP—large-scale atomistic simulation with neural network potential, EANN—embedded atom neural network, AIMNet—atoms-in-molecules Net, PIP—permutationally invariant polynomial, PaiNN—polarizable atom interaction neural network, RANN—rapid artificial neural network, NequIP—neural equivariant interatomic potential, NEP—Neuro evolution potential, GemNet—geometric message passing neural network, PFP—PreFerred potential, ml-ACE—multi-layer atomic cluster expansion, BIGDML—Bravais-inspired gradient domain machine learning, REANN—recursively embedded atom neural network, HermNet—heterogeneous relational message passing network, BOTNet—body-ordered-tensor-network, ETNs—equivalent tensor networks, PESPIP—potential energy surface PIP, GNoME—graph networks for materials exploration, EDDP—ephemeral data derived potential, CHGNet—crystal Hamiltonian graph neural network, ALIGNN-FF—atomistic line graph neural network-based force field, AIMNet2—2nd generation of atoms-in-molecules neural network potential, ViSNet—vector-scalar interactive graph neural network, SEGNN—spindistance edge graph neural network, MB-PIPNet—ManyBody PIPNet, HotPP—high-order tensor message passing interatomic potential, GPTFF—graph-based pre-trained transformer force field, EquiREANN—equivalent REANN, CAMP—Cartesian atomic moment machine learning interatomic potential, CACE—Cartesian atomic cluster expansion)
在第一类基于局域结构描述符的模型中,基于对称多项式构建的模型因其数学上的完备性和形式上的简洁性而备受关注。其中,MTP模型采用一组完备的对称多项式对原子环境进行描述,可视为传统多项式不变势(permutationally invariant polynomial,PIP)模型在局域描述框架下的重要发展。相较于其他常见的MLFF模型,MTP模型在精度与效率之间取得了优异的平衡。Han等[59]在C和S体系中,比较了包含2体、3体项的余弦特征(MTP、ACSF、Bispectrum、DP-Chebyshev、DP-Gaussian及ACE等描述符),发现在采用相同描述符长度和线性回归模型的条件下,MTP描述符所构建的线性模型拟合精度最优。进一步,Zuo等[60]针对Ni、Li、Si、Cu、Mo、Ge等多种材料体系,系统评估了MTP、NNP、GAP、SNAP、qSNAP等主流势函数模型的精度及计算效率,评估结果如图3[60]所示。可见,MTP在保持同等精度的前提下,具备更优的计算效率,其数据点紧贴Pareto边界,展现出模型精度与计算效率的优异平衡。值得强调的是,MTP模型与其他多项式形式的模型存在深刻的联系。Drautz[24]和Dusson等[61]证明了MTP模型与ACE模型之间存在等价性。除此之外,MTP模型也可以看作Hodapp和Shapeev[62]提出的等变张量网络(equivariant tensor network,ETN)模型的特殊形式。
图3
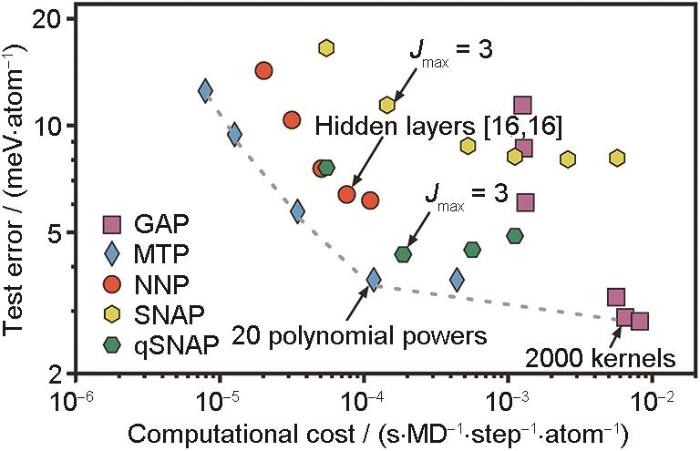
本文聚焦Shapeev[21]提出的MTP势函数模型,回顾了MTP势函数模型的理论基础,介绍了基于MTP的模型发展,以及该模型在一些典型材料科学体系中的应用,最后对MTP模型未来的发展方向进行展望。
1 矩张量模型
式中,
式中,
为解决张量形式下的参考坐标系依赖性问题,Shapeev[21]提出通过张量缩并构造旋转不变的标量基组
式中,
式中,
从上面讨论可以看到,
式中,
2 矩张量模型的优化和改进
式中,
图4

需特别说明的是,
针对上述问题,Wang等[66]采用遗传算法为每个标量函数确定最优分解路径。具体实施步骤如下:首先,为每个
式中,
式中,
3 基于主动学习的训练集生成
下面简要介绍一下D-最优化准则。对于具有线性回归形式的势函数,将
图5

图5
结合主动学习采样方法的矩张量势(MTP)模型开发流程
Fig.5
Workflow of developing a (MTP) model combined with active learning sampling method (γ(x*)—extrapolation grade for configuration x*, γthr—grade threshold)
图6

图6
MTP模型在QM9数据集上的主动学习与随机选择策略的效率和性能比较[64]
Fig.6
Efficiency and property comparisons of active learning vs random sampling for training a MTP model on the QM9 dataset[64] (a, b) root-mean-square (RMS) error (a) and maximal error (b) as a function of training set size (c) parity plot of MTP-predicted enthalpy (HMT) vs reference enthalpy (Href) for a model trained with random selection (d) parity plot for a model trained with active learning
4 MTP模型在不同体系中的应用
自提出以来,MTP模型已在材料科学的诸多子领域得到成功验证。本节从结构搜索、金属和合金、固-固相变、表面和界面现象等几个典型方面,系统回顾MTP模型的代表性应用研究,以展示其强大的计算模拟能力和广泛的适用性。
4.1 结构搜索
基于第一性原理的结构搜索受限于计算成本,通常仅能处理至多数十原子规模的体系,而机器学习势函数的应用则有效突破了这一限制。目前,MTP模型已在USPEX[76],MAGUS[77]等结构搜索软件中应用。Podryabinkin等[78]将MTP模型与USPEX软件及主动学习策略相结合,提出了一种动态构建势函数的高效搜索框架。该框架在结构搜索过程中通过主动学习不断优化势函数,并将其应用于单质C、B及高压Na体系的结构探索。该方案能够高效筛选出基态附近的稳定构型,并成功发现了一种由54个原子组成的新型硼晶体结构。Gubaev等[79]将该方案应用于Cu-Pd、Co-Ni-V、Al-Ni-Ti等合金体系,高效地搜索凸包附近的结构,并在Al-Ni-Ti体系中成功预测了AlNi11、Al4Ni8、AlNi9Ti2三种新的热力学稳定结构。
4.2 金属和传统合金
MTP模型已经广泛应用于金属和合金体系的模拟中。Novoselov等[80]利用MTP模型计算了Al、Mo、Si中的空位扩散行为,其结果与密度泛函理论(DFT)结果高度吻合。Shapeev等[81]利用MTP模型研究了bcc-Ti的弹性不变效应。该工作计算了900~1700 K下bcc-Ti的弹性常数,发现该温度区间内bcc-Ti弹性模量的温度依赖性极弱,展现出单质金属中独特的Elinvar效应(弹性不变效应)。Mukhamedov等[82]在
对于以W为代表的难熔元素,其在高温下强烈的晶格非谐性对传统模拟方法构成挑战。Forslund和Ruban[84]采用基于Langevin动力学的两阶段升采样热力学积分(TU-TILD)方法,利用MTP作为中间模型,以DFT精度实现了金属W七个不同晶面在温度达到熔点前的表面自由能的高效计算。计算结果表明,对于更开放的表面,非谐效应在3000 K时显著增强,致使表面能各向异性随温度升高而减弱,而最密排(110)表面非谐性较弱。计算得到的Wulff形状和表面能数据在接近熔点时与实验结果高度吻合。Forslund等[85]采用基于上述方法改进的直接升采样方法进一步研究了V、Ta、Mo和W四种bcc结构难熔金属直至熔点的热容、热胀系数、体模量等热力学性质。结果表明,这四种元素电子贡献(电子热激发及电子-振动耦合)及非谐效应贡献均十分显著,并可结合电子结构计算阐释第V族和VI族难熔金属的热力学性质变化规律。基于熔点构建的同源温度标度分析表明,计算得到的同源温度依赖性与实验数据高度吻合,证明了采用MTP作为中间模型的直接升采样算法在第一性原理精度的热力学性质计算方面的能力。Srinivasan等[86]同样利用MTP作为中间模型,采用直接升采样方法计算了Nb、Mo、Ta和W四种难熔金属的显式非谐自由能和熵值。结果表明,四种元素的显式非谐性差异并非源于高温行为,而是源自其基态状态,即温度效应会使得Mo和W的bcc结构失稳,从而对于Nb和Ta则起到稳定作用。Zhu等[87]利用MTP模型结合TU-TILD方法研究了V、W及无序VW合金的熔化特性,发现热振动对电子态密度具有显著影响,进而显著改变电子自由能贡献。该效应在W中表现明显,在V中则被组态熵抵消,在VW合金中该效应较W稍弱。该结果表明,电子自由能贡献对于难熔合金热力学性质影响不可忽视。Jung等[88]采用相同的方法,以DFT精度计算了fcc结构的Nb、Ni、Al及hcp-Mg单质基态至熔点的平衡态热力学性质。Zhang等[89]提出了用于计算包含热激发效应的空位扩散过渡态Gibbs自由能的过渡态热力学积分(TSTI)方案,并采用MTP模型作为全非谐性的模型,研究了bcc-W中非谐性对温度依赖的空位形成和迁移Gibbs自由能的影响,计算结果与实验结果高度吻合,成功解释了W自扩散非Arrhenius行为的物理起源。上述研究均采用了升采样方法[90],其中MTP模型以其精度和效率兼备的特点,成为该类型方案中中间参考态计算的不二之选[91]。
合金中的缺陷对其性能具有关键影响。因此,势函数能否精确模拟这些缺陷的性质,是评估其有效性和可靠性的核心指标。Zotov等[92]构建了bcc-Nb体系的MTP模型,该模型不仅精确复现了DFT计算的位错核心结构及位错迁移的Peierls能垒,还精确描述了位错易核能量及临界分切应力(CRSS)的孪生-反孪生不对称性,充分展示了MTP模型对位错缺陷的建模能力。在辐照损伤模拟方面,Wang等[93]针对bcc-Fe的辐照损伤问题设计了混合MTP模型。该模型通过光滑的切换函数,将适用于极短程碰撞的Ziegler-Biersack-Littmark (ZBL)核排斥势、描述短程相互作用的MTP-S势以及描述中长程相互作用的MTP-L势进行了无缝的跨尺度耦合,实现了从高能级联碰撞到缺陷演化的全过程精确模拟。Kwon等[94]采用MTP和路径积分分子动力学(PIMD)模拟方法,系统研究了三种bcc金属(Nb、Fe和W)中的H扩散行为。在合理温度范围内,其计算的扩散系数和观测到的同位素效应均与实验数据高度吻合,证实了MTP模型在准确表征bcc金属中的H扩散动力学方面的可靠性。Kumar等[95]利用MTP模型研究了H在C15立方及C14六方TiCr2H x (
Wang等[66]针对Ni-Al二元体系,采用两阶段主动学习策略构建了高精度的MTP势函数模型,该模型使用优化后的MTP基组,在保持高精度的同时也具备极高的计算效率。该模型可以高精度地预测Ni及Ni3Al相的弹性常数、各类点缺陷及点缺陷团簇形成能/结合能、界面、广义层错能、声子谱等基本物理性质,且预测精度显著优于现有的EAM等经验势函数。基于此势函数,该工作系统研究了镍合金中不同大小团簇的稳定构型及空位团簇形成能/结合能随团簇大小的变化关系,并研究了堆垛层错四面体(SFT)类型空位团簇的扩散过程。分子动力学(MD)模拟结果表明,六空位SFT在小空位团簇中具有更高的稳定性和更高的迁移速率,并表征了SFT迁移原子尺度上的动力学过程(图7c[66])。Xu等[96]研究了Ni3Al中的反常屈服行为,利用MTP模型高效地计算了包含非简谐效应的复杂堆垛层错(CSF)滑移的自由能面,发现了Ni3Al中交滑移过程能垒随温度升高而增大的现象,并揭示了该现象主要来自非谐振动(图7a[96])和纵向的自旋涨落。Xu等[97]进一步考虑了Ni3Al体系的屈服应力反常现象,利用MTP势函数研究了Ni3Al中的位错行为,观测并分析了Kear-Wilsdorf锁(KWL)的形成与解锁过程(图7b[97]),发现其解锁应力呈现显著的温度依赖性。该发现与现有解析模型中普遍认为解锁过程非温度依赖的基本假设相矛盾,不仅为理解KWL的形成和解锁机制提供了新的原子尺度视角,也为揭示屈服应力反常现象的物理起源提供了更深入的理论依据,同时充分展现了MTP模型在描述复杂位错核心结构和热激活过程方面的强大能力。
图7

图7
MTP模型在Ni-Al合金体系中的应用[66,96,97]
Fig.7
Applications of MTP model on Ni-Al alloys
(a) distributions of projected first-nearest-neighboring pairs for both the complex stacking fault (CSF) and bulk Ni3Al at 1454 K[96] (d—displacement)
(b) temperature-dependent critical stresses to unlock a Kear-Wilsdorf lock (KWL)[97] (τ1, τ2—critical stresses for unlocking the KWL)
(c) migration of stacking fault tetrahedra (SFT) cluster in Ni[66]
式中,
在先前的案例中,电子自由能仍需通过DFT计算得到。进一步,Srinivasan等[102]将电子温度效应纳入了MTP框架中,将MTP模型扩展为eMTP模型。在eMTP框架下,
式中,
4.3 高熵合金
高熵合金(HEA)因其近乎无限的成分空间而具有广阔的性质可调控性,但这种复杂性使得原子间势函数的开发极具挑战性。本节针对MTP模型在高熵合金体系的应用(图8[103~105])进行综述。Gubaev等[103]对比了Gaussian矩神经网络(GM-NN)与MTP模型在TaVCrW体系中的性能,在成分、构型及振动自由等多个方面展开分析(图8a[103]),发现两者具有相当的预测能力,但MTP模型在训练规模收敛速率方面表现更优。Jafary-zadeh等[106]采用MTP模型揭示了Co-Fe-Ni三元多主元合金中,静态/动态晶格畸变、热膨胀及化学短程有序(SRO)对弹性性能的耦合影响机制。Gubaev等[107]利用MTP模型研究了不同Ta浓度下,bcc-TiZrHfTa x 体系在有限温度下的结构-弹性相关关系,发现合金弹性性能与结构
图8

Fig.8
Applications of MTP model in HEAs
(a) RMS errors in predicted energies and forces for the MTP model evaluated on the test data for different in-distribution subsystems of Ta-V-Cr-W[103]
(b) scale and plasticity mechanism-dependent strengths of MoNbTaW and W in polycrystal and single-crystalline nanopillar[104] (dc, dc1-dc3—critical values; MPEA—multi-principal element alloy)
(c) kink nucleation mechanism and the observation of cross-slip locking[105] (d110—interplanar spacing of the (110) plane)
难熔高熵合金(RHEA)具有优异的高温强度,合金中刃型位错和螺型位错对其塑性变形具有重要影响。Yin等[105]利用MTP模型系统研究了bcc-MoNbTaW RHEA中刃型和螺型位错在宽温域内的迁移机制及SRO对这些机制的影响规律,发现螺位错的交叉滑移锁定机制是RHEA强化作用的关键(图8c[105]),SRO的存在会促进刃型位错的迁移,但抑制螺型位错运动中的双扭折形核速率,而螺型位错运动中的交滑移锁定机制与SRO无关。该工作展示了MTP模型对复杂合金位错缺陷良好的描述能力。Song等[110]采用MTP模型研究了钛基RHEAs在有限温度下的弹性性能,发现软化声子模式是导致钛基RHEAs弹性常数、模量异常以及各向异性随温度升高的主要原因。
4.4 固-固相变
MTP模型在固-固相变领域也获得了广泛应用,如图9[111~113]所示。Jung等[114]采用MTP模型结合直接升采样算法,实现了体积-温度相空间稳定区域的高效扫描,并在稳定区间内的密集网格上进行热力学积分。利用该方案,Jung等[114]计算了Ti、Zr、Hf体系中hcp-bcc相变特性及两相的热力学性质。在Ti3O5体系中,Liu等[111]利用结构因子作为集合变量(collective variable,CV),结合增强采样与主动学习技术构建了完备的MTP,并借此研究了β-Ti3O5至λ-Ti3O5的超快可逆重构型相变过程。该工作采用巨分子动力学(metadynamics)方法计算了两相的温压相图,预测在0 GPa下相变温度为535 K,与实验值(470 K)高度吻合。Liu等[111]还观察到,β相及λ相沿c方向的任意的乐高式堆垛结构均具有动力学稳定性(图9a[111])。图9b[111]为对自由能面的计算。可见,相对于协同转变,逐层转变在动力学上更具有优势。在拉伸应变条件下的大尺度直接MD模拟中观察到了层内转变的面内形核长大过程,进一步的静态能垒计算表明,面内转变更倾向于形核机制,故而β-λ相变为面内形核、多步逐层转变的能垒降低的过程。该研究[111]不仅阐明了具体的相变路径,更展示了MTP结合增强采样技术在探索复杂重构型相变自由能面方面的巨大潜力。Liu等[112]利用MTP模型研究了Van der Waals层状材料In2Se3的多晶型相变过程,构建了In2Se3体系热力学相图(图9c[112]),并系统研究了该体系中
图9

Fig.9
Applications of MTP model on solid-solid phase transitions
(a) lego-like metastable phases in Ti3O5[111] (c and a—lattice constants)
(b) free energy as a function of collective variables during
(c) phase diagram of In2Se3[112]
(d) domain wall configurations in
(e) vibrational, orientational, and total entropy of low-temperature monoclinic (M-I), intermediate-temperature monoclinic (M-II), fcc (C), and high-pressure rhombohedral (R) phases of KPF6 as a function of temperature[113] (Sori—orientational contribution, Svib—vibrational contribution, Stot—total contribution)
4.5 表界面体系
MTP模型在金属表面分子吸附及表面化学领域也展现出突出的应用潜力。Chen等[116]结合MD模拟与增强采样技术,构建了Cu表面溶剂模型的MTP函数,精确预测了CO*、OH*、COH*、HCO*及OCCHO*在Cu表面的吸附能,以及乙二醇在Cu和Pd表面发生C—H键断裂所需的自由能势垒,这些结果与DFT结果高度吻合,展现出了MTP模型在表面催化问题中的应用潜力。Klimanova等[117]利用MTP模型结合主动学习算法,系统研究了CO/Pd(111)、NO/Pd(111)、NH3/Cu(100)、C6H6/Ag(111)及CH2CO/Rh(211)表面吸附体系的稳定吸附位点,结果与DFT计算结果相吻合。
在表界面催化研究中,过渡金属表面CO吸附是一个经典谜题(the CO puzzle),即传统的DFT计算会错误预测过渡金属表面CO的稳定吸附位点,并系统性地低估表面能、高估吸附能[118,119]。尽管多体无规相近似(RPA)方法不仅能给出与定性实验一致的结果,还能给出准确的定量预测[120],但是该方法因十分昂贵而无法用于大体系、高通量计算。为此,Liu等[121]针对Rh(0001)表面CO吸附问题,利用MTP模型结合差分机器学习(Δ-learning)算法,在低成本下构建了具备RPA精度的机器学习势函数模型,成功预测了不同CO覆盖度下的吸附行为。如图10a[121]所示,基于RPA构建的MLFF能准确预测Rh(111)表面能、CO吸附位点偏好以及不同覆盖度下的吸附能,预测结果均与实验数据高度吻合。此外,Liu等[121]还预测了覆盖度依赖的基态吸附构型及吸附饱和覆盖度13/16 ML (monolayer),即表面吸附位点中13/16被占据。该工作利用MTP作为沟通高精度RPA计算与大规模计算模拟的桥梁,通过Δ-learning策略将RPA级别的精度“迁移”到高效的MTP势函数上,解决了低成本下高精度势函数开发“老大难”的问题。
图10

图10
MTP模型在表面及界面体系中的应用[121,122,125]
Fig.10
Applications of MTP model on surface and interface systems
(a) CO adsorption/binding energies and adsorption configurations on Rh(0001) surface[121] (ML—monolayer, PBE—Perdew-Burke-Ernzerhof functional theory, RPA—random phase approximation, vdW-DF—Van der Waals density functional theory, MTP-RPAΔ—MTP with RPA accuracy generated by the
(b) Arrhenius plot of Li areal density at the interfacial regions Li-S cathode interface[125] (Ea—Li-Fe diffusion energy barrier, D—diffusivity)
(c) free energy profile for the protons along the intermolecular axes, and snapshots from the path integral molecular dynamics (PIMD) trajectory representing proton transfer process[122] (δ—reaction coordinate, R
H2O分子在Ru(0001)表面是否存在解离是一个争论已久的问题,许多实验观测和理论计算都给出了不同的结论。Cao等[122]基于优化的MTP模型针对该体系开发了高效的MLFF,并采用该模型在大超胞体系上进行了纳秒级PIMD模拟,重新审视了H2O分子在Ru(0001)表面是否解离的问题。结果表明,核量子效应导致的质子量子离域化促使H2O分子间发生快速、频繁的质子转移,进而促进H2O分子在Ru(0001)表面的解离,PIMD观测到的质子转移能垒及水解离过程见图10c[122]。该工作为Ru(0001)表面H2O分子解离现象提供了直接理论证据,为H2O/金属界面的原子尺度机理研究提供了关键见解[122]。
Li6PS5X (X = Cl、Br、I)是一种全固态锂离子电池中固态电解质的有力候选材料,Ou等[123]构建了Li6PS5Cl体系的MTP函数。计算结果表明,倾斜晶界
5 总结与展望
本文系统梳理了矩张量机器学习势模型的数学理论基础和模型发展历程,详细阐述了基于MTP的主动学习方法,并全面综述了矩张量机器学习势在单质、合金、相变材料、表面/界面等多元材料体系模拟中的应用进展。尽管MTP在精度与效率之间展现出优良的平衡性,其进一步发展仍面临若干关键挑战,具体体现在以下方面。
(1) 模型表达能力与计算成本的权衡。MTP模型作为对称多项式模型,其模型复杂度存在上限,相较于DP、SchNet、MACE等基于神经网络/图神经网络的复杂模型,MTP模型的全相空间描述能力存在不足。尽管引入更高阶的矩张量、更多的径向基组、更高的多项式幂次可以有效拓展MTP模型的描述能力,但是计算成本急剧增加,使得MTP模型的优势不复存在。针对此问题,未来可通过优化获得描述性更强的标量基组,并耦合等变神经网络[48]等具有高数据效率的非线性回归模型,在保留MTP物理解释性的同时突破线性模型表达瓶颈。这一技术路线有望高效处理非晶态材料、高熵合金及复杂有机/无机界面等化学环境高度异质的体系。另一条技术路线为结合MTP的高效特性与通用大模型(如MACE-MP-0模型[57])的泛化能力,进而构建“大模型迁移到小模型”的研究范式:通过大模型获得体系结构的能量、力、应力等高质量训练数据,进而训练专用MTP函数进行大规模、长时程模拟。该方法有望形成“泛化筛选-精准模拟”的新研究框架,为特定复杂材料体系的深度研究提供高效解决方案。
(3) 并行计算架构适配不足。随着计算模拟领域对算力需求的不断提升,图形处理器(GPU)、张量处理器(TPU)、深算单元(DCU)等大规模并行计算设备正日益成为主流硬件平台。尽管MTP模型在中央处理器(CPU)架构上已展现出卓越的计算性能,但其在GPU等加速设备上的移植和优化仍显不足。特别是在张量分量的计算过程中,尚未充分利用GPU的并行计算特性,存在显著的性能提升空间。因此,开发面向现代并行计算架构的高效实现方案,已成为推动MTP模型持续发展的重要方向。
参考文献
Perspective: Materials informatics and big data: Realization of the “fourth paradigm” of science in materials science
[J].
A review of computational methods in materials science: Examples from shock-wave and polymer physics
[J].
This review discusses several computational methods used on different length and time scales for the simulation of material behavior. First, the importance of physical modeling and its relation to computer simulation on multiscales is discussed. Then, computational methods used on different scales are shortly reviewed, before we focus on the molecular dynamics (MD) method. Here we survey in a tutorial-like fashion some key issues including several MD optimization techniques. Thereafter, computational examples for the capabilities of numerical simulations in materials research are discussed. We focus on recent results of shock wave simulations of a solid which are based on two different modeling approaches and we discuss their respective assets and drawbacks with a view to their application on multiscales. Then, the prospects of computer simulations on the molecular length scale using coarse-grained MD methods are covered by means of examples pertaining to complex topological polymer structures including star-polymers, biomacromolecules such as polyelectrolytes and polymers with intrinsic stiffness. This review ends by highlighting new emerging interdisciplinary applications of computational methods in the field of medical engineering where the application of concepts of polymer physics and of shock waves to biological systems holds a lot of promise for improving medical applications such as extracorporeal shock wave lithotripsy or tumor treatment.
Embedded-atom method: Derivation and application to impurities, surfaces, and other defects in metals
[J].
A simple empirical N-body potential for transition metals
[J].
Modified embedded-atom potentials for cubic materials and impurities
[J].
New empirical approach for the structure and energy of covalent systems
[J].
Neural network models of potential energy surfaces
[J].
Combining ab initio computations, neural networks, and diffusion Monte Carlo: An efficient method to treat weakly bound molecules
[J].We describe a new method to calculate the vibrational ground state properties of weakly bound molecular systems and apply it to (HF)2 and HF–HCl. A Bayesian Inference neural network is used to fit an analytic function to a set of ab initio data points, which may then be employed by the quantum diffusion Monte Carlo method to produce ground state vibrational wave functions and properties. The method is general and relatively simple to implement and will be attractive for calculations on systems for which no analytic potential energy surface exists.
Representation of intermolecular potential functions by neural networks
[J].
The fitting of potential energy surfaces using neural networks: Application to the study of vibrational levels of
Ab initio potential energy and dipole moment surfaces for
Using neural networks to represent potential surfaces as sums of products
[J].By using exponential activation functions with a neural network (NN) method we show that it is possible to fit potentials to a sum-of-products form. The sum-of-products form is desirable because it reduces the cost of doing the quadratures required for quantum dynamics calculations. It also greatly facilitates the use of the multiconfiguration time dependent Hartree method. Unlike potfit product representation algorithm, the new NN approach does not require using a grid of points. It also produces sum-of-products potentials with fewer terms. As the number of dimensions is increased, we expect the advantages of the exponential NN idea to become more significant.
Generalized neural-network representation of high-dimensional potential-energy surfaces
[J].
Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons
[J].
The gaussian approximation potential: An interatomic potential derived from first principles quantum mechanics
[D].
On representing chemical environments
[J].
On-the-fly machine learning force field generation: Application to melting points
[J].
Phase transitions of hybrid perovskites simulated by machine-learning force fields trained on the fly with Bayesian inference
[J].
Moment tensor potentials: A class of systematically improvable interatomic potentials
[J].
Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials
[J].
Extending the accuracy of the SNAP interatomic potential form
[J].The Spectral Neighbor Analysis Potential (SNAP) is a classical interatomic potential that expresses the energy of each atom as a linear function of selected bispectrum components of the neighbor atoms. An extension of the SNAP form is proposed that includes quadratic terms in the bispectrum components. The extension is shown to provide a large increase in accuracy relative to the linear form, while incurring only a modest increase in computational cost. The mathematical structure of the quadratic SNAP form is similar to the embedded atom method (EAM), with the SNAP bispectrum components serving as counterparts to the two-body density functions in EAM. The effectiveness of the new form is demonstrated using an extensive set of training data for tantalum structures. Similar to artificial neural network potentials, the quadratic SNAP form requires substantially more training data in order to prevent overfitting. The quality of this new potential form is measured through a robust cross-validation analysis.
Atomic cluster expansion for accurate and transferable interatomic potentials
[J].
LASP: Fast global potential energy surface exploration
[J].Here we introduce the LASP code, which is designed for large‐scale atomistic simulation of complex materials with neural network (NN) potential. The software architecture and functionalities of LASP will be overviewed. LASP features with the global neural network (G‐NN) potential that is generated by learning the first principles dataset of global PES from stochastic surface walking (SSW) global optimization. The combination of the SSW method with global NN potential facilitates greatly the PES exploration for a wide range of complex materials. Not limited to SSW‐NN global optimization, the software implements standard interfaces to dock with other energy/force evaluation packages and can also perform common tasks for computing PES properties, such as single‐ended and double‐ended transition state search, the molecular dynamics simulation with and without restraints. A few examples are given to illustrate the efficiency and capabilities of LASP code. Our ongoing efforts for code developing and G‐NN potential library building are also presented.
Recent implementations in LASP 3.0: Global neural network potential with multiple elements and better long-range description
[J].LASP (large-scale atomistic simulation with neural network potential) software developed by our group since 2018 is a powerful platform (www.lasphub.com) for performing atomic simulation of complex materials. The software integrates the neural network (NN) potential technique with the global potential energy surface exploration method, and thus can be utilized widely for structure prediction and reaction mechanism exploration. Here we introduce our recent update on the LASP program version 3.0, focusing on the new functionalities including the advanced neural network training based on the multi-network framework, the newly-introduced S7 and S8 power type structure descriptor (PTSD). These new functionalities are designed to further improve the accuracy of potentials and accelerate the neural network training for multiple-element systems. Taking Cu-C-H-O neural network potential and a heterogeneous catalytic model as the example, we show that these new functionalities can accelerate the training of multi-element neural network potential by using the existing single-network potential as the input. The obtained double-network potential CuCHO is robust in simulation and the introduction of S7 and S8 PTSDs can reduce the root-mean-square errors of energy by a factor of two.
Glucose to 5-hydroxymethylfurfural: Origin of site-selectivity resolved by machine learning based reaction sampling
[J].
Dynamic coordination of cations and catalytic selectivity on zinc-chromium oxide alloys during syngas conversion
[J].
Atomic structure of boron resolved using machine learning and global sampling
[J].
Material discovery by combining stochastic surface walking global optimization with a neural network
[J].
High-dimensional neural network potentials for magnetic systems using spin-dependent atom-centered symmetry functions
[J].Machine learning potentials have emerged as a powerful tool to extend the time and length scales of first-principles quality simulations. Still, most machine learning potentials cannot distinguish different electronic spin arrangements and thus are not applicable to materials in different magnetic states. Here we propose spin-dependent atom-centered symmetry functions as a type of descriptor taking the atomic spin degrees of freedom into account. When used as an input for a high-dimensional neural network potential (HDNNP), accurate potential energy surfaces of multicomponent systems can be constructed, describing multiple collinear magnetic states. We demonstrate the performance of these magnetic HDNNPs for the case of manganese oxide, MnO. The method predicts the magnetically distorted rhombohedral structure in excellent agreement with density functional theory and experiment. Its efficiency allows to determine the Néel temperature considering structural fluctuations, entropic effects, and defects. The method is general and is expected to be useful also for other types of systems such as oligonuclear transition metal complexes.
Four generations of high-dimensional neural network potentials
[J].Since their introduction about 25 years ago, machine learning (ML) potentials have become an important tool in the field of atomistic simulations. After the initial decade, in which neural networks were successfully used to construct potentials for rather small molecular systems, the development of high-dimensional neural network potentials (HDNNPs) in 2007 opened the way for the application of ML potentials in simulations of large systems containing thousands of atoms. To date, many other types of ML potentials have been proposed continuously increasing the range of problems that can be studied. In this review, the methodology of the family of HDNNPs including new recent developments will be discussed using a classification scheme into four generations of potentials, which is also applicable to many other types of ML potentials. The first generation is formed by early neural network potentials designed for low-dimensional systems. High-dimensional neural network potentials established the second generation and are based on three key steps: first, the expression of the total energy as a sum of environment-dependent atomic energy contributions; second, the description of the atomic environments by atom-centered symmetry functions as descriptors fulfilling the requirements of rotational, translational, and permutation invariance; and third, the iterative construction of the reference electronic structure data sets by active learning. In third-generation HDNNPs, in addition, long-range interactions are included employing environment-dependent partial charges expressed by atomic neural networks. In fourth-generation HDNNPs, which are just emerging, in addition, nonlocal phenomena such as long-range charge transfer can be included. The applicability and remaining limitations of HDNNPs are discussed along with an outlook at possible future developments.
A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer
[J].Machine learning potentials have become an important tool for atomistic simulations in many fields, from chemistry via molecular biology to materials science. Most of the established methods, however, rely on local properties and are thus unable to take global changes in the electronic structure into account, which result from long-range charge transfer or different charge states. In this work we overcome this limitation by introducing a fourth-generation high-dimensional neural network potential that combines a charge equilibration scheme employing environment-dependent atomic electronegativities with accurate atomic energies. The method, which is able to correctly describe global charge distributions in arbitrary systems, yields much improved energies and substantially extends the applicability of modern machine learning potentials. This is demonstrated for a series of systems representing typical scenarios in chemistry and materials science that are incorrectly described by current methods, while the fourth-generation neural network potential is in excellent agreement with electronic structure calculations.
Deep potential: A general representation of a many-body potential energy surface
[J].
DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics
[J].
Advances in machine learning molecular dynamics to assist materials nucleation and solidification research
[J].Solidification nucleation is an everlasting research topic in the fields of materials science and condensed matter physics. Molecular dynamics (MD) and enhanced sampling methods provide a powerful means to observe the microscopic mechanisms of solidification processes in situ at the atomic level and to analyze the thermodynamic and kinetic properties of phase transitions. Recent advancements in MD simulations, particularly those incorporating machine learning (ML) techniques, have remarkably advanced our understanding of nucleation across different systems. This paper first reviews the basic theory of solidification nucleation and introduces common methods used in solidification nucleation simulation studies. It then delves into the application of ML techniques in three key areas: force fields, enhanced sampling, and order parameters. The paper further highlights several representative systems to demonstrate the practical applications of these methods. Finally, a summary and outlook on the future of ML-assisted MD simulations for studying material solidification were provided.
机器学习分子动力学辅助材料凝固形核研究进展
[J].凝固形核是材料科学、凝聚态物理等领域长盛不衰的研究热点。分子动力学以及增强采样方法为从原子尺度原位观察凝固过程微观机理、解析相变热力学与动力学性质提供了有力手段。近年来,该领域的研究者们开发了一些融合机器学习技术的先进分子动力学模拟新方法,在多种体系的形核研究中取得了一定成果。本文首先回顾了凝固形核的基本理论,并从势函数、增强采样、形核序参量3个方面介绍凝固形核模拟研究中的常用方法以及机器学习技术在其中的应用。然后,选取了几个具有代表性的体系并介绍相关方法的实际应用。最后,对机器学习分子动力学辅助材料凝固形核模拟研究领域进行了总结与展望。
Application of machine learning force fields for modeling ferroelectric materials
[J].Ferroelectric materials, which are characterized by tunable spontaneous polarization, show remarkable application potential for nonvolatile information storage; however they present various challenges. The performance of these materials is strongly influenced by their dynamic polarization behavior under multiple external fields. Due to the limited precision of experimental observations, precise atomic-level material simulations are crucial. Although molecular dynamics (MD) offers an ideal method for investigating material dynamics over a wide spatiotemporal range, its application to new materials is often limited by challenges such as low accuracy, complex development, and limited portability of conventional classical force fields. Advances in machine learning have provided new possibilities for developing force fields. Among different machine learning potentials, deep potential (DP) based on deep neural networks stands out. DP offers accuracy comparable to that of density functional theory while providing computational efficiency similar to that of conventional classical force fields. This review primarily focused on the development and application of DP in ferroelectric materials, specifically examining the phase transition mechanisms and polarization reversal processes at the atomic scale. Considerable efforts have been made to develop and evaluate DP for crucial ferroelectric materials such as hafnium dioxide (HfO2) and classic perovskite ferroelectric materials. Furthermore, this review explores the high oxygen-ion migration kinetics in HfO2 using DP and investigates the flexoelectricity induced by polar domain boundaries and the bulk photovoltaic effects in strontium titanate. By highlighting the use of DP molecular dynamics approaches in ferroelectric materials, this review emphasizes the role of machine learning approaches in optimizing and accelerating material simulations to facilitate further breakthroughs and discoveries.
机器学习势在铁电材料研究中的应用
[J].
Deep potentials for materials science
[J].
Neural message passing for quantum chemistry
[A].
SchNet—A deep learning architecture for molecules and materials
[J].Deep learning has led to a paradigm shift in artificial intelligence, including web, text, and image search, speech recognition, as well as bioinformatics, with growing impact in chemical physics. Machine learning, in general, and deep learning, in particular, are ideally suitable for representing quantum-mechanical interactions, enabling us to model nonlinear potential-energy surfaces or enhancing the exploration of chemical compound space. Here we present the deep learning architecture SchNet that is specifically designed to model atomistic systems by making use of continuous-filter convolutional layers. We demonstrate the capabilities of SchNet by accurately predicting a range of properties across chemical space for molecules and materials, where our model learns chemically plausible embeddings of atom types across the periodic table. Finally, we employ SchNet to predict potential-energy surfaces and energy-conserving force fields for molecular dynamics simulations of small molecules and perform an exemplary study on the quantum-mechanical properties of C20-fullerene that would have been infeasible with regular ab initio molecular dynamics.
PhysNet: A neural network for predicting energies, forces, dipole moments, and partial charges
[J].In recent years, machine learning (ML) methods have become increasingly popular in computational chemistry. After being trained on appropriate ab initio reference data, these methods allow for accurately predicting the properties of chemical systems, circumventing the need for explicitly solving the electronic Schrödinger equation. Because of their computational efficiency and scalability to large data sets, deep neural networks (DNNs) are a particularly promising ML algorithm for chemical applications. This work introduces PhysNet, a DNN architecture designed for predicting energies, forces, and dipole moments of chemical systems. PhysNet achieves state-of-the-art performance on the QM9, MD17, and ISO17 benchmarks. Further, two new data sets are generated in order to probe the performance of ML models for describing chemical reactions, long-range interactions, and condensed phase systems. It is shown that explicitly including electrostatics in energy predictions is crucial for a qualitatively correct description of the asymptotic regions of a potential energy surface (PES). PhysNet models trained on a systematically constructed set of small peptide fragments (at most eight heavy atoms) are able to generalize to considerably larger proteins like deca-alanine (Ala): The optimized geometry of helical Ala predicted by PhysNet is virtually identical to ab initio results (RMSD = 0.21 Å). By running unbiased molecular dynamics (MD) simulations of Ala on the PhysNet-PES in gas phase, it is found that instead of a helical structure, Ala folds into a "wreath-shaped" configuration, which is more stable than the helical form by 0.46 kcal mol according to the reference ab initio calculations.
Directional message passing for molecular graphs
[A].
Equivariant message passing for the prediction of tensorial properties and molecular spectra
[J].
E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials
[J].This work presents Neural Equivariant Interatomic Potentials (NequIP), an E(3)-equivariant neural network approach for learning interatomic potentials from ab-initio calculations for molecular dynamics simulations. While most contemporary symmetry-aware models use invariant convolutions and only act on scalars, NequIP employs E(3)-equivariant convolutions for interactions of geometric tensors, resulting in a more information-rich and faithful representation of atomic environments. The method achieves state-of-the-art accuracy on a challenging and diverse set of molecules and materials while exhibiting remarkable data efficiency. NequIP outperforms existing models with up to three orders of magnitude fewer training data, challenging the widely held belief that deep neural networks require massive training sets. The high data efficiency of the method allows for the construction of accurate potentials using high-order quantum chemical level of theory as reference and enables high-fidelity molecular dynamics simulations over long time scales.© 2022. The Author(s).
Learning local equivariant representations for large-scale atomistic dynamics
[J].A simultaneously accurate and computationally efficient parametrization of the potential energy surface of molecules and materials is a long-standing goal in the natural sciences. While atom-centered message passing neural networks (MPNNs) have shown remarkable accuracy, their information propagation has limited the accessible length-scales. Local methods, conversely, scale to large simulations but have suffered from inferior accuracy. This work introduces Allegro, a strictly local equivariant deep neural network interatomic potential architecture that simultaneously exhibits excellent accuracy and scalability. Allegro represents a many-body potential using iterated tensor products of learned equivariant representations without atom-centered message passing. Allegro obtains improvements over state-of-the-art methods on QM9 and revMD17. A single tensor product layer outperforms existing deep MPNNs and transformers on QM9. Furthermore, Allegro displays remarkable generalization to out-of-distribution data. Molecular simulations using Allegro recover structural and kinetic properties of an amorphous electrolyte in excellent agreement with ab-initio simulations. Finally, we demonstrate parallelization with a simulation of 100 million atoms.© 2023. The Author(s).
MACE: Higher order equivariant message passing neural networks for fast and accurate force fields
[J].
A simple and efficient equivariant message-passing neural network model for non-local potential energy surface
[J].
The design space of E(3)-equivariant atom-centered interatomic potentials
[J].
Latent Ewald summation for machine learning of long-range interactions
[J].
Commentary: The materials project: A materials genome approach to accelerating materials innovation
[J].
Graph networks as a universal machine learning framework for molecules and crystals
[J].Graph networks are a new machine learning (ML) paradigm that supports both relational reasoning and combinatorial generalization. Here, we develop universal MatErials Graph Network (MEGNet) models for accurate property prediction in both molecules and crystals. We demonstrate that the MEGNet models outperform prior ML models such as the SchNet in 11 out of 13 properties of the QM9 molecule data set. Similarly, we show that MEGNet models trained on similar to 60 000 crystals in the Materials Project substantially outperform prior ML models in the prediction of the formation energies, band gaps, and elastic moduli of crystals, achieving better than density functional theory accuracy over a much larger data set. We present two new strategies to address data limitations common in materials science and chemistry. First, we demonstrate a physically intuitive approach to unify four separate molecular MEGNet models for the internal energy at 0 K and room temperature, enthalpy, and Gibbs free energy into a single free energy MEGNet model by incorporating the temperature, pressure, and entropy as global state inputs. Second, we show that the learned element embeddings in MEGNet models encode periodic chemical trends and can be transfer-learned from a property model trained on a larger data set (formation energies) to improve property models with smaller amounts of data (band gaps and elastic moduli).
A universal graph deep learning interatomic potential for the periodic table
[J].Interatomic potentials (IAPs), which describe the potential energy surface of atoms, are a fundamental input for atomistic simulations. However, existing IAPs are either fitted to narrow chemistries or too inaccurate for general applications. Here we report a universal IAP for materials based on graph neural networks with three-body interactions (M3GNet). The M3GNet IAP was trained on the massive database of structural relaxations performed by the Materials Project over the past ten years and has broad applications in structural relaxation, dynamic simulations and property prediction of materials across diverse chemical spaces. About 1.8 million materials from a screening of 31 million hypothetical crystal structures were identified to be potentially stable against existing Materials Project crystals based on M3GNet energies. Of the top 2,000 materials with the lowest energies above the convex hull, 1,578 were verified to be stable using density functional theory calculations. These results demonstrate a machine learning-accelerated pathway to the discovery of synthesizable materials with exceptional properties.© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling
[J].
Scaling deep learning for materials discovery
[J].
Pretraining of attention-based deep learning potential model for molecular simulation
[J].
DPA-2: A large atomic model as a multi-task learner
[J].
A foundation model for atomistic materials chemistry
[J].
GPTFF: A high-accuracy out-of-the-box universal AI force field for arbitrary inorganic materials
[J].This study introduces a novel artificial intelligence (AI) force field, namely a graph-based pre-trained transformer force field (GPTFF), which can simulate arbitrary inorganic systems with good precision and generalizability. Harnessing a large trove of the data and the attention mechanism of transformer algorithms, the model can accurately predict energy, atomic force, and stress with mean absolute error (MAE) values of 32 meV/atom, 71 meV/Å, and 0.365 GPa, respectively. The dataset used to train the model includes 37.8 million single-point energies, 11.7 billion force pairs, and 340.2 million stresses. We also demonstrated that the GPTFF can be universally used to simulate various physical systems, such as crystal structure optimization, phase transition simulations, and mass transport. The model is publicly released with this paper, enabling anyone to use it immediately without needing to train it.Copyright © 2024 Science China Press. Published by Elsevier B.V. All rights reserved.
Accuracy evaluation of different machine learning force field features
[J].
A performance and cost assessment of machine learning interatomic potentials
[J].
Atomic cluster expansion: Completeness, efficiency and stability
[J].
Equivariant tensor network potentials
[J].
Active learning of linearly parametrized interatomic potentials
[J].
Machine learning of molecular properties: Locality and active learning
[J].
The MLIP package: Moment tensor potentials with MPI and active learning
[J].
Efficient moment tensor machine-learning interatomic potential for accurate description of defects in Ni-Al Alloys
[J].
Active learning with statistical models
[J].For many types of machine learning algorithms, one can compute the statistically `optimal' way to select training data. In this paper, we review how optimal data selection techniques have been used with feedforward neural networks. We then show how the same principles may be used to select data for two alternative, statistically-based learning architectures: mixtures of Gaussians and locally weighted regression. While the techniques for neural networks are computationally expensive and approximate, the techniques for mixtures of Gaussians and locally weighted regression are both efficient and accurate. Empirically, we observe that the optimality criterion sharply decreases the number of training examples the learner needs in order to achieve good performance.
On-the-fly active learning of interatomic potentials for large-scale atomistic simulations
[J].The on-the-fly generation of machine-learning force fields by active-learning schemes attracts a great deal of attention in the community of atomistic simulations. The algorithms allow the machine to self-learn an interatomic potential and construct machine-learned models on the fly during simulations. State-of-the-art query strategies allow the machine to judge whether new structures are out of the training data set or not. Only when the machine judges the necessity of updating the data set with the new structures are first-principles calculations carried out. Otherwise, the yet available machine-learned model is used to update the atomic positions. In this manner, most of the first-principles calculations are bypassed during training, and overall, simulations are accelerated by several orders of magnitude while retaining almost first-principles accuracy. In this Perspective, after describing essential components of the active-learning algorithms, we demonstrate the power of the schemes by presenting recent applications.
Machine-learned potentials for next-generation matter simulations
[J].The choice of simulation methods in computational materials science is driven by a fundamental trade-off: bridging large time- and length-scales with highly accurate simulations at an affordable computational cost. Venturing the investigation of complex phenomena on large scales requires fast yet accurate computational methods. We review the emerging field of machine-learned potentials, which promises to reach the accuracy of quantum mechanical computations at a substantially reduced computational cost. This Review will summarize the basic principles of the underlying machine learning methods, the data acquisition process and active learning procedures. We highlight multiple recent applications of machine-learned potentials in various fields, ranging from organic chemistry and biomolecules to inorganic crystal structure predictions and surface science. We furthermore discuss the developments required to promote a broader use of ML potentials, and the possibility of using them to help solve open questions in materials science and facilitate fully computational materials design.
Escaping free-energy minima
[J].We introduce a powerful method for exploring the properties of the multidimensional free energy surfaces (FESs) of complex many-body systems by means of coarse-grained non-Markovian dynamics in the space defined by a few collective coordinates. A characteristic feature of these dynamics is the presence of a history-dependent potential term that, in time, fills the minima in the FES, allowing the efficient exploration and accurate determination of the FES as a function of the collective coordinates. We demonstrate the usefulness of this approach in the case of the dissociation of a NaCl molecule in water and in the study of the conformational changes of a dialanine in solution.
Simulating rare events in equilibrium or nonequilibrium stochastic systems
[J].
Well-tempered metadynamics: A smoothly converging and tunable free-energy method
[J].
A brief review of integrated tempering sampling molecular simulation
[J].
Machine learning assisted canonical sampling (MLACS)
[J].
Quantum chemistry structures and properties of 134 kilo molecules
[J].
USPEX—Evolutionary crystal structure prediction
[J].
MAGUS: Machine learning and graph theory assisted universal structure searcher
[J].Crystal structure predictions based on first-principles calculations have gained great success in materials science and solid state physics. However, the remaining challenges still limit their applications in systems with a large number of atoms, especially the complexity of conformational space and the cost of local optimizations for big systems. Here, we introduce a crystal structure prediction method, MAGUS, based on the evolutionary algorithm, which addresses the above challenges with machine learning and graph theory. Techniques used in the program are summarized in detail and benchmark tests are provided. With intensive tests, we demonstrate that on-the-fly machine-learning potentials can be used to significantly reduce the number of expensive first-principles calculations, and the crystal decomposition based on graph theory can efficiently decrease the required configurations in order to find the target structures. We also summarized the representative applications of this method on several research topics, including unexpected compounds in the interior of planets and their exotic states at high pressure and high temperature (superionic, plastic, partially diffusive state, etc.); new functional materials (superhard, high-energy-density, superconducting, photoelectric materials), etc. These successful applications demonstrated that MAGUS code can help to accelerate the discovery of interesting materials and phenomena, as well as the significant value of crystal structure predictions in general.
Accelerating crystal structure prediction by machine-learning interatomic potentials with active learning
[J].
Accelerating high-throughput searches for new alloys with active learning of interatomic potentials
[J].
Moment tensor potentials as a promising tool to study diffusion processes
[J].
Elinvar effect in β-Ti simulated by on-the-fly trained moment tensor potential
[J].A combination of quantum mechanics calculations with machine learning techniques can lead to a paradigm shift in our ability to predict materials properties from first principles. Here we show that on-the-fly training of an interatomic potential described through moment tensors provides the same accuracy as state-of-the-art ab initio molecular dynamics in predicting high-temperature elastic properties of materials with two orders of magnitude less computational effort. Using the technique, we investigate high-temperature bcc phase of titanium and predict very weak, Elinvar, temperature dependence of its elastic moduli, similar to the behavior of the so-called GUM Ti-based alloys (Sato et al 2003 Science \n 300 464). Given the fact that GUM alloys have complex chemical compositions and operate at room temperature, Elinvar properties of elemental bcc-Ti observed in the wide temperature interval 1100–1700 K is unique.
Machine learning interatomic potential for the low-modulus Ti-Nb-Zr alloys in the vicinity of dynamical instability
[J].
Development of machine learning interatomic potential for zinc
[J].
Ab initio surface free energies of tungsten with full account of thermal excitations
[J].
Thermodynamic properties on the homologous temperature scale from direct upsampling: Understanding electron-vibration coupling and thermal vacancies in bcc refractory metals
[J].
Anharmonicity in bcc refractory elements: A detailed ab initio analysis
[J].
Melting properties of the refractory metals V and W and the binary VW alloy fully from first principles
[J].
High-accuracy thermodynamic properties to the melting point from ab initio calculations aided by machine-learning potentials
[J].
Ab initio machine-learning unveils strong anharmonicity in non-Arrhenius self-diffusion of tungsten
[J].The knowledge of diffusion mechanisms in materials is crucial for predicting their high-temperature performance and stability, yet accurately capturing the underlying physics like thermal effects remains challenging. In particular, the origin of the experimentally observed non-Arrhenius diffusion behavior has remained elusive, largely due to the lack of effective computational tools. Here we propose an efficient ab initio framework to compute the Gibbs energy of the transition state in vacancy-mediated diffusion including the relevant thermal excitations at the density-functional-theory level. With the aid of a bespoke machine-learning interatomic potential, the temperature-dependent vacancy formation and migration Gibbs energies of the prototype system body-centered cubic (BCC) tungsten are shown to be strongly affected by anharmonicity. This finding explains the physical origin of the experimentally observed non-Arrhenius behavior of tungsten self-diffusion. A remarkable agreement between the calculated and experimental temperature-dependent self-diffusivity and, in particular, its curvature is revealed. The proposed computational framework is robust and broadly applicable, as evidenced by first tests for a hexagonal close-packed (HCP) multicomponent high-entropy alloy. The successful applications underscore the attainability of an accurate ab initio diffusion database.
Improved method of calculating ab initio high-temperature thermodynamic properties with application to ZrC
[J].
Ab initio vibrational free energies including anharmonicity for multicomponent alloys
[J].The unique and unanticipated properties of multiple principal component alloys have reinvigorated the field of alloy design and drawn strong interest across scientific disciplines. The vast compositional parameter space makes these alloys a unique area of exploration by means of computational design. However, as of now a method to compute efficiently, yet with high accuracy the thermodynamic properties of such alloys has been missing. One of the underlying reasons is the lack of accurate and efficient approaches to compute vibrational free energies—including anharmonicity—for these chemically complex multicomponent alloys. In this work, a density-functional-theory based approach to overcome this issue is developed based on a combination of thermodynamic integration and a machine-learning potential. We demonstrate the performance of the approach by computing the anharmonic free energy of the prototypical five-component VNbMoTaW refractory high entropy alloy.
Moment tensor potential for static and dynamic investigations of screw dislocations in bcc Nb
[J].
Machine-learning interatomic potential for radiation damage effects in bcc-iron
[J].
Accurate description of hydrogen diffusivity in bcc metals using machine-learning moment tensor potentials and path-integral methods
[J].
Machine learning potentials for hydrogen absorption in TiCr2 Laves phases
[J].
Accurate complex-stacking-fault Gibbs energy in Ni3Al at high temperatures
[J].
Origin of the yield stress anomaly in L12 intermetallics unveiled with physically informed machine-learning potentials
[J].
Magnetic Moment Tensor Potentials for collinear spin-polarized materials reproduce different magnetic states of bcc Fe
[J].We present the magnetic Moment Tensor Potentials (mMTPs), a class of machine-learning interatomic potentials, accurately reproducing both vibrational and magnetic degrees of freedom as provided, e.g., from first-principles calculations. The accuracy is achieved by a two-step minimization scheme that coarse-grains the atomic and the spin space. The performance of the mMTPs is demonstrated for the prototype magnetic system bcc iron, with applications to phonon calculations for different magnetic states, and molecular-dynamics simulations with fluctuating magnetic moments.
Constrained DFT-based magnetic machine-learning potentials for magnetic alloys: a case study of Fe-Al
[J].We propose a machine-learning interatomic potential for multi-component magnetic materials. In this potential we consider magnetic moments as degrees of freedom (features) along with atomic positions, atomic types, and lattice vectors. We create a training set with constrained DFT (cDFT) that allows us to calculate energies of configurations with non-equilibrium (excited) magnetic moments and, thus, it is possible to construct the training set in a wide configuration space with great variety of non-equilibrium atomic positions, magnetic moments, and lattice vectors. Such a training set makes possible to fit reliable potentials that will allow us to predict properties of configurations in the excited states (including the ones with non-equilibrium magnetic moments). We verify the trained potentials on the system of bcc Fe-Al with different concentrations of Al and Fe and different ways Al and Fe atoms occupy the supercell sites. Here, we show that the formation energies, the equilibrium lattice parameters, and the total magnetic moments of the unit cell for different Fe-Al structures calculated with machine-learning potentials are in good correspondence with the ones obtained with DFT. We also demonstrate that the theoretical calculations conducted in this study qualitatively reproduce the experimentally-observed anomalous volume-composition dependence in the Fe-Al system.© 2023. The Author(s).
Fitting to magnetic forces improves the reliability of magnetic moment tensor potentials
[J].
Constitution and magnetic properties of iron-rich iron-aluminum alloys
[J].
Electronic moment tensor potentials include both electronic and vibrational degrees of freedom
[J].We present the electronic moment tensor potentials (eMTPs), a class of machine-learning interatomic models and a generalization of the classical MTPs, reproducing both the electronic and vibrational degrees of freedom, up to the accuracy of ab initio calculations. Following the original polynomial interpolation idea of the MTPs, the eMTPs are defined as polynomials of vibrational and electronic degrees of freedom, corrected to have a finite interatomic cutoff. Practically, an eMTP is constructed from the classical MTPs fitted to a training set, whose energies and forces are calculated with electronic temperatures corresponding to the Chebyshev nodes on a given temperature interval. The eMTP energy is hence a Chebyshev interpolation of the classical MTPs. Using the eMTP, one can obtain the temperature-dependent vibrational free energy including anharmonicity coming from phonon interactions, the electronic free energy coming from electron interactions, and the coupling of atomic vibrations and electronic excitations. Each of the contributions can be accessed individually using the proposed formalism. The performance of eMTPs is demonstrated for two refractory systems which have a significant electronic, vibrational and coupling contribution up to the melting point—unary Nb, and a disordered TaVCrW high-entropy alloy. Highly accurate thermodynamic and kinetic quantities can now be obtained just by using eMTPs, without any further ab initio calculations. The proposed construction to include the electronic degree of freedom can also be applied to other machine-learning models.
Performance of two complementary machine-learned potentials in modelling chemically complex systems
[J].\n Chemically complex multicomponent alloys possess exceptional properties derived from an inexhaustible compositional space. The complexity however makes interatomic potential development challenging. We explore two complementary machine-learned potentials—the moment tensor potential (MTP) and the Gaussian moment neural network (GM-NN)—in simultaneously describing configurational\n and\n vibrational degrees of freedom in the Ta-V-Cr-W alloy family. Both models are equally accurate with excellent performance evaluated against density-functional-theory. They achieve root-mean-square-errors (RMSEs) in energies of less than a few meV/atom across 0 K ordered and high-temperature disordered configurations included in the training. Even for compositions not in training, relative energy RMSEs at high temperatures are within a few meV/atom. High-temperature molecular dynamics forces have similarly small RMSEs of about 0.15 eV/Å for the disordered quaternary included in, and ternaries not part of training. MTPs achieve faster convergence with training size; GM-NNs are faster in execution. Active learning is partially beneficial and should be complemented with conventional human-based training set generation.\n
Size-dependent strength superiority in multi-principal element alloys versus constituent metals: Insights from machine-learning atomistic simulations
[J].
Atomistic simulations of dislocation mobility in refractory high-entropy alloys and the effect of chemical short-range order
[J].Refractory high-entropy alloys (RHEAs) are designed for high elevated-temperature strength, with both edge and screw dislocations playing an important role for plastic deformation. However, they can also display a significant energetic driving force for chemical short-range ordering (SRO). Here, we investigate mechanisms underlying the mobilities of screw and edge dislocations in the body-centered cubic MoNbTaW RHEA over a wide temperature range using extensive molecular dynamics simulations based on a highly-accurate machine-learning interatomic potential. Further, we specifically evaluate how these mechanisms are affected by the presence of SRO. The mobility of edge dislocations is found to be enhanced by the presence of SRO, whereas the rate of double-kink nucleation in the motion of screw dislocations is reduced, although this influence of SRO appears to be attenuated at increasing temperature. Independent of the presence of SRO, a cross-slip locking mechanism is observed for the motion of screws, which provides for extra strengthening for refractory high-entropy alloy system.© 2021. The Author(s).
Applying a machine learning interatomic potential to unravel the effects of local lattice distortion on the elastic properties of multi-principal element alloys
[J].
Finite-temperature interplay of structural stability, chemical complexity, and elastic properties of bcc multicomponent alloys from ab initio trained machine-learning potentials
[J].
Machine-learning potentials enable predictive and tractable high-throughput screening of random alloys
[J].
Accelerating ab initio melting property calculations with machine learning: Application to the high entropy alloy TaVCrW
[J].Melting properties are critical for designing novel materials, especially for discovering high-performance, high-melting refractory materials. Experimental measurements of these properties are extremely challenging due to their high melting temperatures. Complementary theoretical predictions are, therefore, indispensable. One of the most accurate approaches for this purpose is the ab initio free-energy approach based on density functional theory (DFT). However, it generally involves expensive thermodynamic integration using ab initio molecular dynamic simulations. The high computational cost makes high-throughput calculations infeasible. Here, we propose a highly efficient DFT-based method aided by a specially designed machine learning potential. As the machine learning potential can closely reproduce the ab initio phase-space distribution, even for multi-component alloys, the costly thermodynamic integration can be fully substituted with more efficient free energy perturbation calculations. The method achieves overall savings of computational resources by 80% compared to current alternatives. We apply the method to the high-entropy alloy TaVCrW and calculate its melting properties, including the melting temperature, entropy and enthalpy of fusion, and volume change at the melting point. Additionally, the heat capacities of solid and liquid TaVCrW are calculated. The results agree reasonably with the CALPHAD extrapolated values.
Machine learning accelerated study on temperature dependent elastic properties of Ti-based refractory high entropy alloys
[J].
Layer-by-layer phase transformation in Ti3O5 revealed by machine-learning molecular dynamics simulations
[J].Reconstructive phase transitions involving breaking and reconstruction of primary chemical bonds are ubiquitous and important for many technological applications. In contrast to displacive phase transitions, the dynamics of reconstructive phase transitions are usually slow due to the large energy barrier. Nevertheless, the reconstructive phase transformation from β- to λ-Ti3O5 exhibits an ultrafast and reversible behavior. Despite extensive studies, the underlying microscopic mechanism remains unclear. Here, we discover a kinetically favorable in-plane nucleated layer-by-layer transformation mechanism through metadynamics and large-scale molecular dynamics simulations. This is enabled by developing an efficient machine learning potential with near first-principles accuracy through an on-the-fly active learning method and an advanced sampling technique. Our results reveal that the β−λ phase transformation initiates with the formation of two-dimensional nuclei in the a\n b-plane and then proceeds layer-by-layer through a multistep barrier-lowering kinetic process via intermediate metastable phases. Our work not only provides important insight into the ultrafast and reversible nature of the β−λ transition, but also presents useful strategies and methods for tackling other complex structural phase transitions.
Diverse polymorphs and phase transitions in van der Waals In2Se3
[J].
Atomistic mechanisms of phase transitions in all-temperature barocaloric material KPF6
[J].
Dynamically stabilized phases with full ab initio accuracy: Thermodynamics of Ti, Zr, Hf with a focus on the hcp-bcc transition
[J].
All-temperature barocaloric effects at pressure-induced phase transitions
[J].
Accelerating explicit solvent models of heterogeneous catalysts with machine learning interatomic potentials
[J].Realistically modelling how solvents affect catalytic reactions is a longstanding challenge due to its prohibitive computational cost. Typically, an explicit atomistic treatment of the solvent molecules is needed together with molecular dynamics (MD) simulations and enhanced sampling methods. Here, we demonstrate the utility of machine learning interatomic potentials (MLIPs), coupled with active learning, to enable fast and accurate explicit solvent modelling of adsorption and reactions on heterogeneous catalysts. MLIPs trained on-the-fly were able to accelerate MD simulations by up to 4 orders of magnitude while reproducing with high fidelity the geometrical features of water in the bulk and at metal-water interfaces. Using these ML-accelerated simulations, we accurately predicted key catalytic quantities such as the adsorption energies of CO*, OH*, COH*, HCO*, and OCCHO* on Cu surfaces and the free energy barriers of C-H scission of ethylene glycol over Cu and Pd surfaces, as validated with calculations. We envision that such simulations will pave the way towards detailed and realistic studies of solvated catalysts at large time- and length-scales.This journal is © The Royal Society of Chemistry.
Accelerating the global search of adsorbate molecule positions using machine-learning interatomic potentials with active learning
[J].We accelerate the global search of adsorbate molecule positions using machine-learning interatomic potentials with active learning.
The CO/Pt(111) puzzle
[J].
CO on Pt(111) puzzle: A possible solution
[J].CO adsorption on the Pt(111) surface is studied using first-principles methods. As found in a recent study [Feibelman et al., J. Phys. Chem. B 105, 4018 (2001)], we find the preferred adsorption site within density functional theory to be the hollow site, whereas experimentally it is found that the top site is preferred. The influence of pseudopotential and exchange-correlation functional error on the CO binding energy and site preference is carefully investigated. We also compare the site preference energy of CO on Pt(111) with the reaction energy of formaldehyde formation from H2 and CO. We show that the discrepancies between the experimental and theoretical results are due to the generalized gradient approximation (GGA) treating different bond orders with varying accuracy. We can therefore expect that GGA results will contain significant error whenever bonds of different bond order are broken and formed.
Random-phase approximation and its applications in computational chemistry and materials science
[J].
Combining machine learning and many-body calculations: Coverage-dependent adsorption of CO on Rh(111)
[J].
Quantum delocalization enables water dissociation on Ru(0001)
[J].
Atomistic modeling of bulk and grain boundary diffusion in solid electrolyte Li6PS5Cl using machine-learning interatomic potentials
[J].
Highly reliable and large-scale simulations of promising argyrodite solid-state electrolytes using a machine-learned moment tensor potential
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}