


Ta的碳化物相比于纯Ta具有更高的强度、硬度、优良的抗烧蚀性能和耐磨性能,工业上一般采用渗碳、化学沉积、等离子喷涂等方式在钽材料表面获得碳化钽层[5~9]。其中渗碳是一种常见的表面热处理工艺,通过渗碳处理可以在Ta及其合金表面形成一层碳化物,能够有效提高材料的使用性能。唐全红等[7]利用真空碳管炉和真空感应炉进行Ta金属渗碳工艺优化,通过调控炉压、渗载比、渗碳时间,成功制备了可耐受2000 ℃高温的短时抗氧化和抗烧蚀渗层。郑欣[8]利用等离子渗碳技术增强钽材料电阻、耐磨和耐腐蚀性能,通过调节渗碳参数能够改变渗层中TaC和 Ta2C的比例,当渗层中TaC为主要成分时,渗层的腐蚀速率较纯Ta可降低100倍以上。周寰林等[9,10]利用离子渗碳在Ta金属表面制备出6 μm的渗碳层,随着渗碳过程的进行,Ta材表面的缺陷逐渐减少,表面层变得更加平整,渗碳后表层硬度提高至原始Ta材的6倍以上,且抗氧化能力显著提升。李佳等[11]利用真空渗碳的方法对金属Ta及Ta-W合金进行渗碳处理,通过对渗碳温度和渗碳时间的优化调控,在Ta基体表面生成含碳化钽的渗层厚度可达40 μm,该工艺能够有效提升Ta基体材料的强度、硬度及抗烧蚀能力,但该工作未对Ta-W合金表面渗碳层进行精细表征。惠鹏飞[12]利用间隙原子渗碳原理对Ta基体进行表面增强处理,在金属Ta表面制备了致密的碳化钽硬质保护层,碳化钽层的显微硬度达到1900 HV,较原始基体提高了13.8倍,有效提升了Ta表面的硬度及耐磨性能。研究者[13,14]对Ta-Mo-C体系和Ta-W-C体系在fcc和hcp之间变化的相图进行了理论研究,分析了Ta-Mo-C、Ta-W-C、Ta-C-N和W-C-N三元体系的相平衡、溶解度和液相线变化。以上研究表明:Ta-Mo和Ta-W合金以及Ta-Mo-C和Ta-W-C体系受到越来越多的关注,而Ta-Mo和Ta-W合金表面渗碳层中的物相组成,特别是Ta的碳化物结构对渗碳层的性能有重要影响,但是利用实验的方法研究Ta的碳化物析出行为及其性能工作量大,时间和经济成本较高。
综上原因,本工作以钽合金为研究对象,构建了含不同合金元素和含量的复合碳化钽(Ta, M)C (M = Mo、W)模型结构,计算了复合碳化钽结构在不同原子组分占比下的能量及其模量、硬度和延展性等力学性能,预测Ta-Mo及Ta-W合金表面复合碳化钽的析出顺序及其性能,通过电子结构分析,探讨复合碳化钽的强化和韧化机制。相关结果可为阐明钽合金表面渗碳层中析出相的析出及形成机制、揭示渗碳层的强韧化机理提供理论指导。
1 计算方法与建立模型
1.1 第一性原理理论计算方法
1.2 能量与力学性质计算方法
式中,
弹性性质是固体材料重要的物理参数,可以通过弹性常数来判断材料的弹性及脆韧性。立方结构有3个独立弹性常数(
式中,Cij 为弹性常数,多晶结构的弹性模量具有更加普遍的应用性,因此本工作中只进行多晶结构模量的计算。
式中,
体积模量(
1.3 结构模型
通过特殊准随机结构(SQS)建模方法,使用合金理论自动化工具包中的mcsqs代码对复合碳化钽分别构建2种晶体结构的超级晶胞[31,32]。其中阴离子位点被C原子占据,阳离子位点按照一定比例的过渡金属原子随机分布。本工作将(Ta, M)C (M = W、Mo)构建为2种晶体结构,如图1a为fcc结构(空间群为Fm
图1
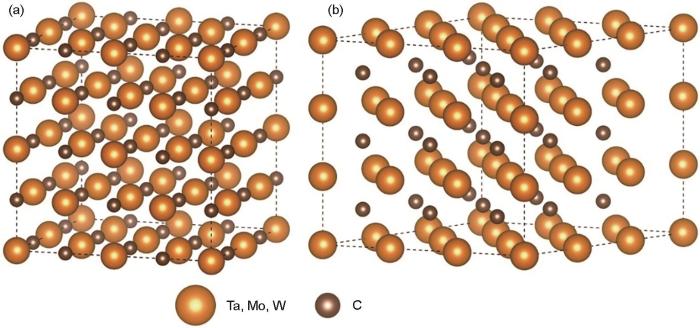
图1
不同结构复合碳化钽的特殊准随机结构(SQS)超胞模型
Fig.1
Super structural models of the special quasi-random structure (SQS) of complex tantalum carbides
(a) fcc (b) hcp
2 计算结果与讨论
2.1 结构稳定性分析
表1为含Mo复合碳化钽结构的晶体参数、内聚能和形成能的具体数据。图2为含Mo复合碳化钽分别在fcc和hcp晶体结构下,不同Mo原子浓度时的内聚能和形成能变化趋势。图2中,hcp晶体结构曲线中的碳化钽为Ta2C,在横坐标中不以Ta2C符号表示,以TaC的符号简略表示(下文所有图中的hcp晶体结构曲线,横坐标碳化钽都以TaC符号简略表示)。由表1可知,fcc相的Ec随着体系中Mo原子浓度的增加而逐渐增大,这意味着具有fcc相结构的含Mo复合碳化钽的稳定性随着Mo浓度的增加而逐渐降低。随着体系中Mo原子浓度的增加,含Mo复合碳化钽的Ef逐渐增大,这表明Mo不利于具有fcc结构的含Mo复合碳化钽的形成。在体系中的Ta被Mo完全取代之前,Ef为负,这表明形成过程在热力学上是有利的。当体系中的Ta完全被Mo取代后,MoC的Ef为正,表明形成过程中存在能量障碍。从表1可以看出,随着体系中Mo原子浓度的增加,hcp相的Ec和Ef逐渐增大,这表明具有hcp结构的含Mo复合碳化钽的稳定性和材料形成能力随着Mo原子浓度的增加而逐渐降低。
表1 含Mo复合碳化钽结构的晶体参数、形成能(Ef)和内聚能(Ec)
Table 1
| Crystal structure | Carbide | Lattice constant nm | Angle (°) | Volume nm3 | Ef eV·atom-1 | Ec eV·atom-1 |
|---|---|---|---|---|---|---|
| fcc | TaC, this work | α = β = γ = 90.00 | 0.71839 | -0.5962 | -8.5998 | |
| TaC[34] | a = 0.447 | -0.59 | ||||
| Ta0.75Mo0.25C | α = 90.04, β = γ = 90.00 | 0.70753 | -0.4089 | -8.1705 | ||
| Ta0.5Mo0.5C | α = β =89.78, γ = 90.22 | 0.69645 | -0.2246 | -7.7442 | ||
| Ta0.25Mo0.75C | α = 89.79, β = γ = 90.00 | 0.68428 | -0.0432 | -7.6155 | ||
| MoC | α = β = γ = 90.00 | 0.66756 | 0.1400 | -6.8956 | ||
| hcp | Ta2C | a = b = 0.31249, c = 0.49593 | α = β = 90.00, γ = 120.00 | 0.33551 | -0.6163 | -8.6566 |
| (P | ||||||
| Ta2C (P | a = b = 0.311, c = 0.495 | γ = 120.00 | -0.60 | |||
| Ta0.75Mo0.25C | a = 0.45271, b = 0.45256, c = 0.43049 | α = 90.05, β = 89.96, γ = 120.01 | 0.68590 | -0.1681 | -7.9208 | |
| Ta0.5Mo0.5C | a = 0.44703, b = 0.44702, c = 0.42883 | α = 90.07, β = 89.98, γ = 120.01 | 0.59361 | -0.1616 | -7.6633 | |
| Ta0.25Mo0.75C | a = 0.44261, b = 0.44227, c = 0.42681 | α = 89.99, β = 90.01, γ = 120.02 | 0.57870 | -0.1610 | -7.4476 | |
| MoC | a = b = 0.43761, c = 0.42422 | α = β = 90.00, γ = 120.00 | 0.56283 | -0.1519 | -7.1876 | |
图2
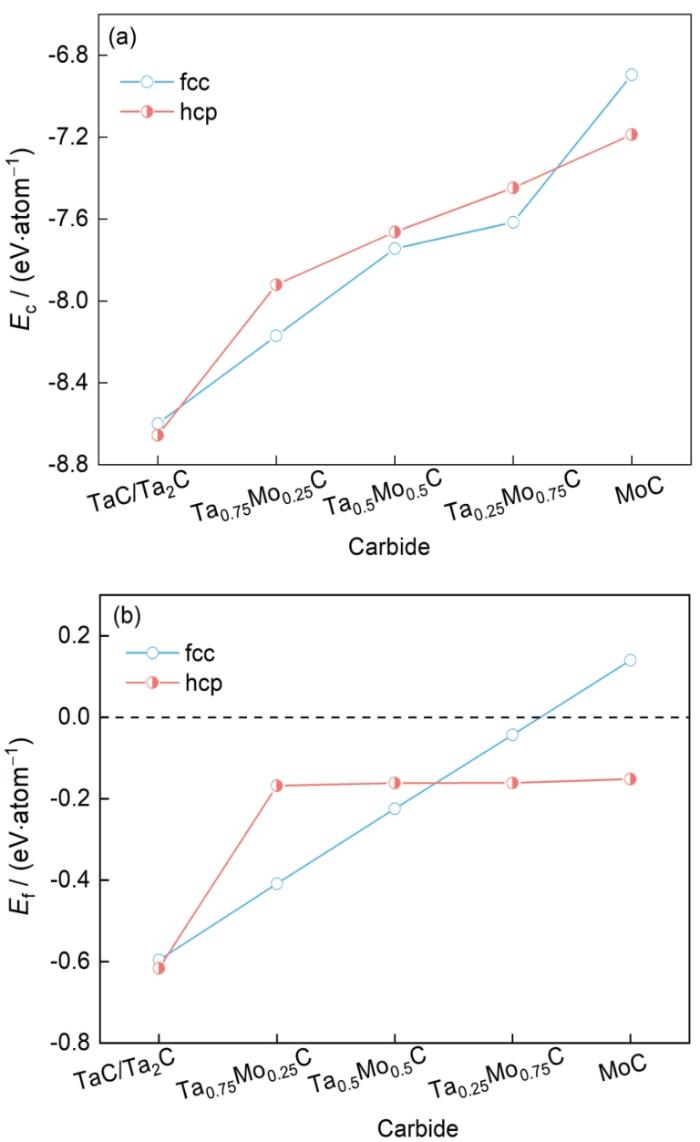
图2
不同结构含Mo复合碳化钽的Ec和Ef
Fig.2
Ec (a) and Ef (b) of complex tantalum carbide containing Mo with different structures
表2为含W复合碳化钽结构的晶体参数、内聚能和形成能。图3给出了fcc和hcp结构含W复合碳化钽内聚能与形成能的关系。从表2可以看出,在TaC中添加25%的W原子会降低系统的Ec。这表明,少量W原子取代了Ta的位置,从而提高了材料的稳定性。W原子浓度的进一步增加会降低体系的Ec,这表明较高浓度的W原子不利于具有fcc相结构的含W复合碳化钽的稳定性。随着体系中W浓度的增加,含W复合碳化钽的Ef也逐渐增加,这表明W不利于形成具有fcc相结构的含W复合碳化钽。W原子浓度在75%及以上时,Ef为正,这意味着形成过程存在能量障碍。从表2可以看出,随着体系中W原子浓度的增加,具有hcp相结构的含W复合碳化钽的Ec先减小后增大,这说明少量的W原子会破坏体系的稳定性,而高浓度的W原子则有利于体系的稳定。随着W原子浓度的增加,体系的Ef逐渐增大,表明形成含W复合碳化钽的能力下降。
表2 含W复合碳化钽结构的晶体参数、Ef和Ec
Table 2
| Crystal structure | Carbide | Lattice constant nm | Angle (°) | Volume nm3 | Ef eV·atom-1 | Ec eV·atom-1 |
|---|---|---|---|---|---|---|
| fcc | TaC, this work | α = β = γ = 90.00 | 0.71839 | -0.5962 | -8.5998 | |
| TaC[34] | a = 0.447 | -0.59 | ||||
| Ta0.75W0.25C | α = 90.00, β = 89.99, γ = 90.00 | 0.70699 | -0.3759 | -8.6364 | ||
| Ta0.5W0.5C | α = 89.79, β = 90.19, γ = 90.06 | 0.69731 | -0.1566 | -8.2123 | ||
| Ta0.25W0.75C | α = β = 90.00, γ = 90.06 | 0.68590 | 0.0565 | -8.3138 | ||
| WC | α = β = γ = 90.00 | 0.67320 | 0.2909 | -7.8169 | ||
| hcp | Ta2C | a = b = 0.31249, c = 0.49593 | α = β = 90.00, γ = 120.00 | 0.33551 | -0.6163 | -8.6566 |
| (P | ||||||
| Ta2C (P | a = b = 0.311, c = 0.495 | γ = 120 | -0.60 | |||
| Ta0.75W0.25C | a = 0.45263, b = 0.45262, c = 0.43127 | α = 89.94, β =90.08, γ = 120.00 | 0.61212 | -0.1640 | -8.1946 | |
| Ta0.5W0.5C | a = 0.44731, b = 0.44718, c = 0.43034 | α = 89.93, β = 89.98, γ = 120.00 | 0.59638 | -0.1561 | -8.2137 | |
| Ta0.25W0.75C | a = 0.44306, b = 0.44305, c = 0.42889 | α = 90.00, β = 89.99, γ = 119.99 | 0.58335 | -0.1573 | -8.2381 | |
| WC | a = b =0.43868, c = 0.42730 | α = β = 90.00, γ = 120.00 | 0.56968 | -0.1543 | -8.2621 | |
图3
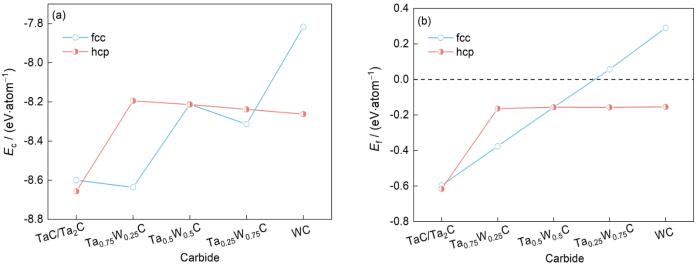
图3
不同结构含W复合碳化钽的Ec和Ef
Fig.3
Energy of complex tantalum carbides containing W with different structures
(a) Ec (b) Ef
以上结果表明,当取代Ta原子的Mo原子和W原子的浓度不超过75%时,具有fcc晶体结构的复合碳化钽更稳定;具有hcp晶体结构的复合碳化钽在Mo/W原子浓度超过75%时更为稳定。其次,当分别取代Ta原子的Mo原子和W原子的浓度不超过50%时,复合碳化钽倾向于形成fcc晶体结构。当浓度超过50%时,复合碳化钽更有可能形成hcp晶体结构。
2.2 力学性能
表3 fcc结构复合碳化钽的弹性常数和力学性质
Table 3
| Carbide (fcc) | C11 ( GPa | C12 ( GPa | C44 ( GPa | G GPa | B GPa | E GPa | HV GPa | G / B | |
|---|---|---|---|---|---|---|---|---|---|
| TaC, this work | 720 | 134 | 154 | 200 | 330 | 500 | 21.8 | 0.607 | 0.25 |
| TaC[34] | 737 | 141 | 175 | 216.90 | 339.67 | 24.53 | |||
| Ta0.75Mo0.25C | 668 | 138 | 152 | 167 | 302 | 423 | 20.9 | 0.605 | 0.25 |
| Ta0.5Mo0.5C | 576 | 165 | 146 | 151 | 310 | 389 | 17.0 | 0.554 | 0.27 |
| Ta0.25Mo0.75C | 581 | 174 | 123 | 122 | 339 | 327 | 13.2 | 0.486 | 0.29 |
| MoC | 684 | 166 | 70 | 200 | 331 | 499 | 7.0 | 0.360 | 0.34 |
| Ta0.75W0.25C | 715 | 139 | 156 | 179 | 310 | 450 | 21.5 | 0.603 | 0.25 |
| Ta0.5W0.5C | 630 | 150 | 147 | 156 | 341 | 407 | 18.8 | 0.577 | 0.26 |
| Ta0.25W0.75C | 611 | 207 | 132 | 129 | 370 | 346 | 12.4 | 0.458 | 0.30 |
| WC | 769 | 170 | 67 | 200 | 330 | 500 | 7.0 | 0.348 | 0.34 |
表4 hcp结构复合碳化钽的弹性常数 (GPa)
Table 4
| Carbide (hcp) | C11 | C12 | C13 | C33 | C44 |
|---|---|---|---|---|---|
| Ta2C (P | 480 | 143 | 138 | 498 | 120 |
| Ta2C (P | 479 | 164 | 149 | 504 | 133 |
| Ta0.75Mo0.25C | 511 | 194 | 138 | 764 | 120 |
| Ta0.5Mo0.5C | 532 | 203 | 151 | 783 | 157 |
| Ta0.25Mo0.75C | 568 | 206 | 159 | 811 | 203 |
| MoC | 618 | 213 | 159 | 854 | 263 |
| Ta0.75W0.25C | 537 | 197 | 138 | 787 | 130 |
| Ta0.5W0.5C | 581 | 209 | 154 | 823 | 181 |
| Ta0.25W0.75C | 621 | 224 | 164 | 878 | 235 |
| WC | 699 | 232 | 167 | 953 | 302 |
表5 hcp结构复合碳化钽的力学性质
Table 5
Carbide (hcp) | G GPa | B GPa | E GPa | Hv GPa | G / B GPa | |
|---|---|---|---|---|---|---|
| Ta2C (P | 149 | 255 | 374 | 16.9 | 0.583 | 0.26 |
| Ta2C (P | 148.08 | 264.66 | 15.87 | |||
| Ta0.75Mo0.25C | 160 | 300 | 407 | 15.6 | 0.532 | 0.27 |
| Ta0.5Mo0.5C | 180 | 315 | 454 | 18.8 | 0.573 | 0.26 |
| Ta0.25Mo0.75C | 209 | 330 | 517 | 23.6 | 0.632 | 0.24 |
| MoC | 246 | 348 | 597 | 30.3 | 0.706 | 0.21 |
| Ta0.75W0.25C | 171 | 310 | 433 | 17.2 | 0.552 | 0.27 |
| Ta0.5W0.5C | 203 | 333 | 507 | 22.1 | 0.609 | 0.25 |
| Ta0.25W0.75C | 234 | 356 | 576 | 26.8 | 0.658 | 0.23 |
| WC | 282 | 385 | 681 | 34.7 | 0.732 | 0.21 |
图4
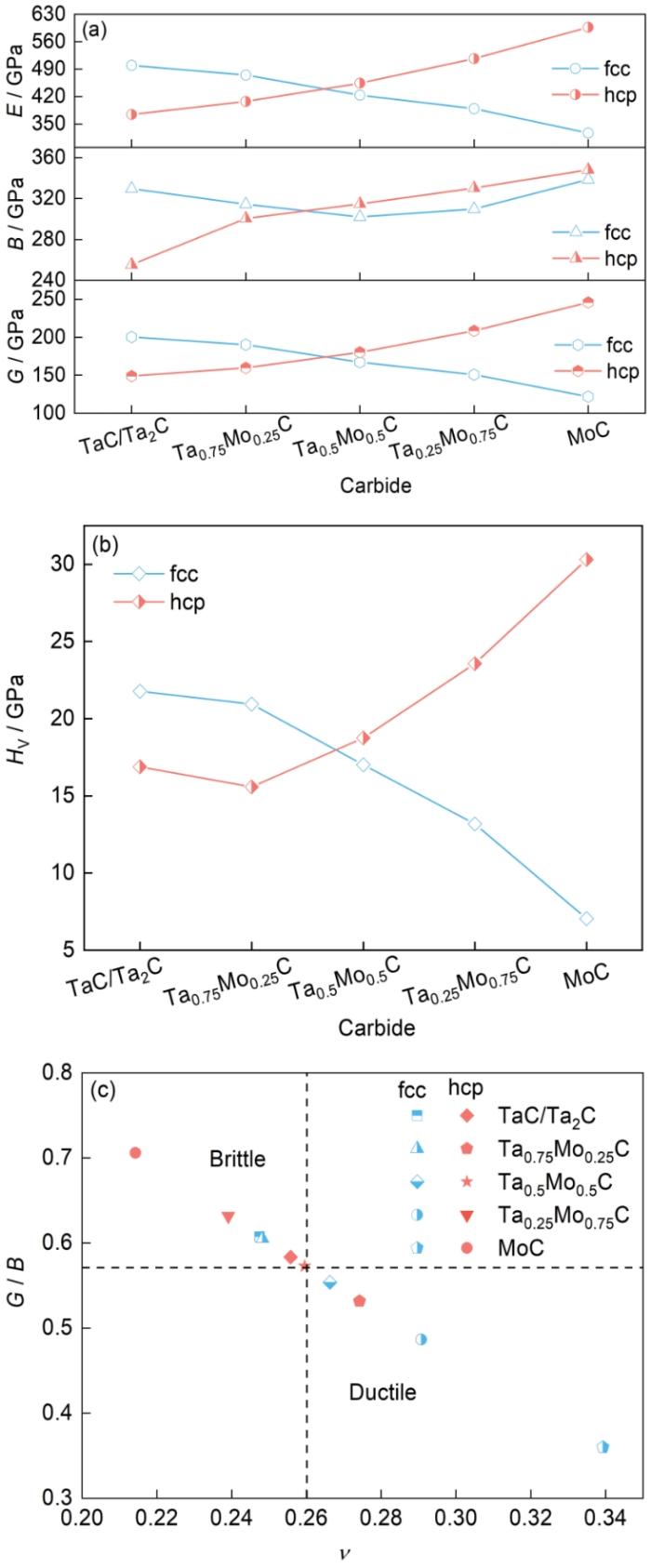
图4
不同结构含Mo复合碳化钽的力学性质
Fig.4
Mechanical properties of complex tantalum carbide containing Mo with different structures
(a) modulus
(b) Vickers hardness
(c) G / Bvs Poisson's ratio
碳化钽的脆性限制了材料的广泛应用。本工作使用G / B和临界Possion比对复合碳化钽的韧性材料和脆性材料进行分类[38]。G / B < 0.571表示材料脆性较大,G / B ≥ 0.571表示材料的延展性较好;
图5
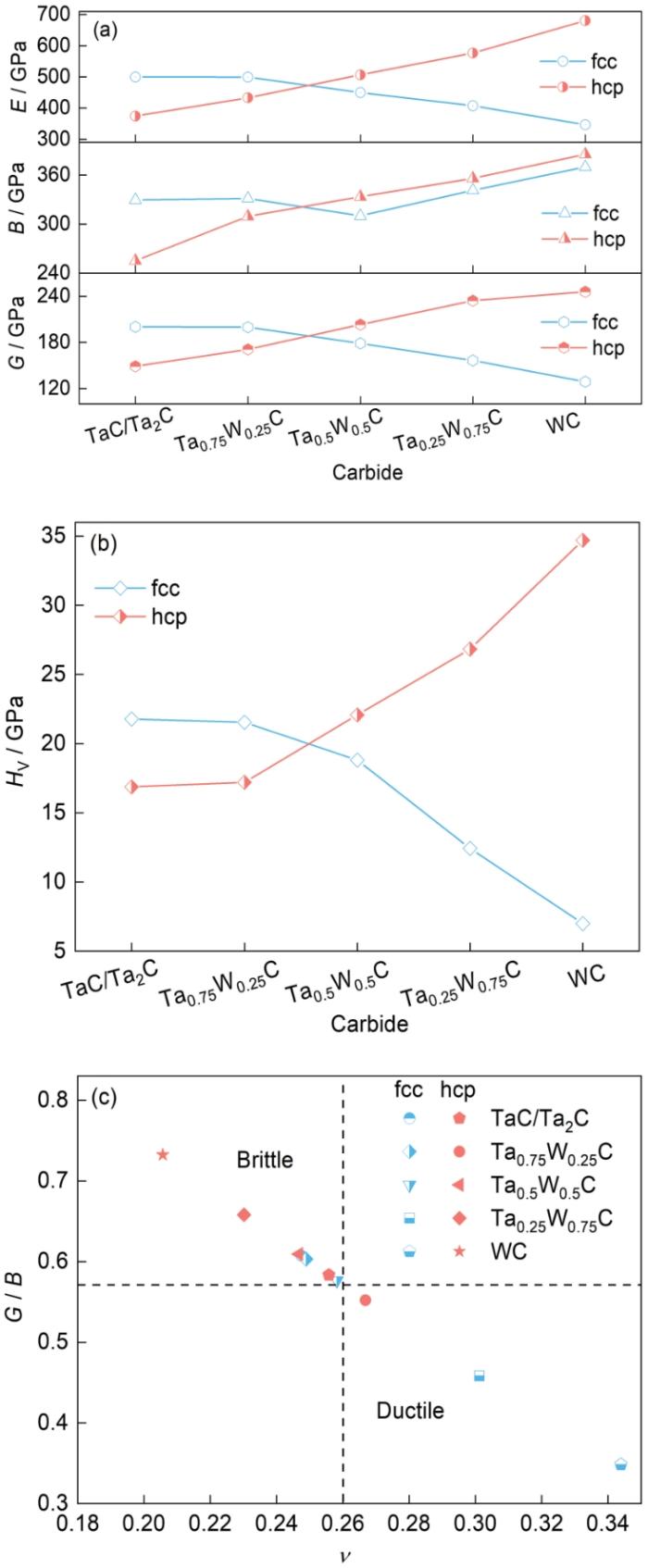
图5
不同结构含W复合碳化钽的力学性质
Fig.5
Mechanical properties of complex tantalum carbide containing W with different structures
(a) modulus
(b) Vickers hardness
(c) G / Bvs Poisson's ratio
综合图4和5的分析结果,不同浓度的Mo原子和W原子会影响复合碳化钽的力学性能。对组成成分相同但晶体结构不同的复合碳化钽也有不同的影响。Mo和W原子的浓度对具有不同晶体结构的复合碳化钽的力学性能具有不同的影响。当体系中Mo原子浓度和W原子浓度小于50%时,具有fcc晶体结构的复合碳化钽具有更高的G、B、E和HV。当Mo和W原子浓度不低于50%时,具有hcp晶体结构的复合碳化钽具有更高的G、B、E和HV。对于具有fcc晶体结构的复合碳化钽,从表3和5、图4c和5c可以看出,随Mo、W的原子浓度增大,
2.3 电子结构分析
为了深入了解复合碳化钽的键合性质,计算了典型复合碳化钽的总态密度(TDOS)和投影态密度(DOS) (图6)。通过研究原子的轨道杂化度,可以有效地表征化学键的离域程度和各原子的轨道杂化性质,有利于分析原子间的成键特征。如图6所示,Fermi能级附近的成键峰主要由Ta5d、Mo4d、W5d和C2p态占据,表明它们对复合碳化钽体系的化学成键有贡献,每种复合碳化钽的总态密度都跨越了Fermi能级,这意味着复合碳化钽能够表现出某些金属特性。在Fermi能级左侧,存在来自金属Ta、Mo或W的d轨道和C的2p轨道之间的杂化重叠,表明Ta、Mo或W金属原子和Cd原子之间存在共价键。在fcc结构中,相比于Mo原子取代,采用W原子取代时,金属和C原子之间的交互作用更强,共价键更强。
图6
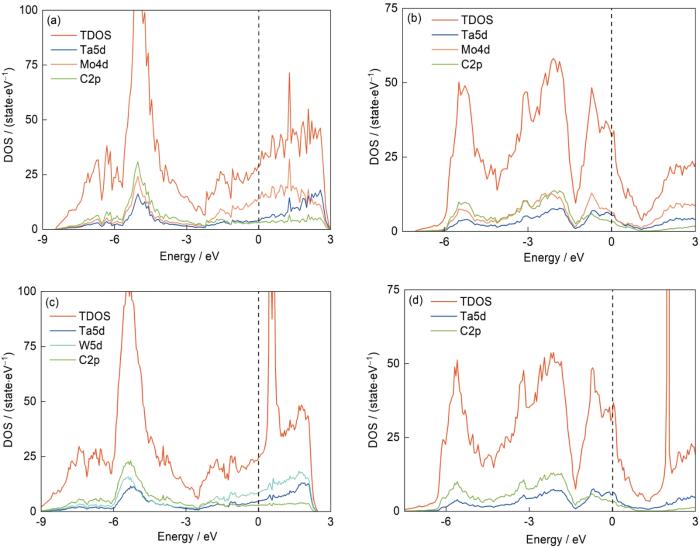
图6
不同结构复合碳化钽的总态密度和投影态密度
Fig.6
Total and projected density of states (DOS) of the same component in multi-component carbides in different crystal structures (The dashed lines indicate the Fermi energy levels; TDOS—total density of states)
(a) Ta0.5Mo0.5C (fcc) (b) Ta0.5Mo0.5C (hcp) (c) Ta0.5W0.5C (fcc) (d) Ta0.5W0.5C (hcp)
式中,
图7为不同结构复合碳化钽fm的变化情况。由图可知,Mo、W原子浓度低于25%时金属键强度降低,Mo、W原子浓度高于25%时金属键强度提高。对比图4c和7a,对于fcc结构的复合碳化钽,fm先减小后增大,复合碳化钽的延展性呈先减小后增大的趋势;对于hcp结构的复合碳化钽,fm先增大后减小,复合碳化钽的延展性呈先增大后减小的趋势。比较图5c和7b,fcc结构的复合碳化钽的fm先减小后增大,延展性同样呈先减小后增大的趋势;具有hcp结构的复合碳化钽的fm先增大后减小,其延展性的变化也是先增大后减小。对上述数据的分析表明,用于描述复合碳化钽金属结合性能的fm与延展性之间存在很强的相关性,复合碳化钽的延展性随着fm的增大而增大。
图7
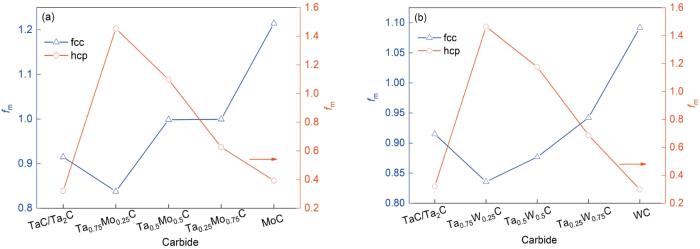
图7
不同结构复合碳化钽的金属丰度(fm)
Fig.7
fm values of complex tantalum carbide containing Mo (a) and W (b) with different structures (fm is an indicator of metal bonding strength)
Mo和W原子的加入有可能改善复合碳化钽的本征脆性。然而,增加Mo和W原子的比例可能会使复合碳化钽结构形成hcp结构。结果表明,Mo和W原子在不同浓度下取代Ta位点时,形成了不同的稳定晶体结构。对于fcc结构的复合碳化钽,Mo和W原子浓度越高,其延展性越好。对于hcp结构的复合碳化钽,少量的Mo和W原子取代Ta位提高了延展性。
表6所示的Bader电荷计算进一步证明了电荷从金属原子转移到C原子,揭示了离子键的作用。表6表明,随着Mo、W原子浓度在复合碳化钽中的增加,离子键强度逐渐降低。由图7和表3可知,Mo、W原子浓度低于25%时金属键强度降低,Mo、W原子浓度高于25%时金属键强度提高;随着Mo、W原子浓度在复合碳化钽中的提高,Poisson比从TaC的0.25逐渐提高到MoC的0.34和WC的0.34,表明复合碳化钽的延展性增强。因此,当Mo、W原子浓度低于25%时,复合碳化钽的离子键强度降低,金属键强度降低,复合碳化钽的延展性不变;当Mo、W原子浓度高于25%时,复合碳化钽的离子键强度降低,金属键强度增强,复合碳化钽的延展性提高。
表6 不同结构复合碳化钽中各原子的平均Bader电荷
Table 6
| Crystal structure | Carbide | C | Ta | Mo | W |
|---|---|---|---|---|---|
| fcc | TaC | 1.7981 | -1.7982 | ||
| Ta0.75Mo0.25C | 1.6594 | -1.8032 | -1.2279 | ||
| Ta0.5Mo0.5C | 1.5107 | -1.7975 | -1.2240 | ||
| Ta0.25Mo0.75C | 1.4359 | -1.8233 | -1.3068 | ||
| MoC | 1.3520 | -1.3520 | |||
| Ta0.75W0.25C | 1.6932 | -1.7852 | -1.4175 | ||
| Ta0.5W0.5C | 1.5430 | -1.5437 | |||
| Ta0.25W0.75C | 1.5538 | -1.8413 | -1.4579 | ||
| WC | 1.5449 | -1.5449 | |||
| hcp | Ta2C | 1.8033 | -0.9016 | ||
| Ta0.75Mo0.25C | 1.4781 | -1.6082 | -1.1061 | ||
| Ta0.5Mo0.5C | 1.3812 | -1.6431 | -1.1380 | ||
| Ta0.25Mo0.75C | 1.3137 | -1.6530 | -1.1950 | ||
| MoC | 1.1998 | -1.1998 | |||
| Ta0.75W0.25C | 1.5063 | -1.6027 | -1.2308 | ||
| Ta0.5W0.5C | 1.4430 | -1.6329 | -1.2666 | ||
| Ta0.25W0.75C | 1.4100 | -1.6492 | -1.3263 | ||
| WC | 1.3852 | -1.3852 |
3 结论
(1) 以Ta-W和Ta-Mo合金为研究对象,构建了含不同合金元素和含量的复合碳化钽(Ta, M)C (M = Mo、W)模型结构。当Mo原子和W原子的浓度不超过75%时,具有fcc结构的复合碳化钽最为稳定。当Mo原子和W原子的浓度超过75%时,六方结构的复合碳化钽最为稳定。当Mo原子和W原子的浓度不超过50%时,复合碳化钽偏向于形成fcc结构。当Mo原子和W原子的浓度超过50%时,复合碳化钽倾向于形成六方结构。
(2) Mo、W原子的加入能够改善复合碳化钽的本征脆性。对于具有fcc结构的复合碳化钽,Mo、W原子浓度的越高,延展性越好;对于具有hcp结构的复合碳化钽,少量Mo、W原子取代Ta原子,有利于提高复合碳化钽的韧性。高浓度Mo、W原子取代Ta原子使复合碳化钽的模量和硬度提高。
(3) 复合碳化钽的成键模式为金属键、共价键和离子键3种键合类型共存,复合碳化钽的延展性变化归因于键合强度的变化,随着金属键强度的增加,(Ta, M)C (M = Mo、W)的延展性增加,离子键强度降低且金属键强度提高是延展性提高的原因。
参考文献
Electrochemical characteristics and impedance spectroscopy studies of carbon-carbon supercapacitors
[J].
A comprehensive review of biological and materials properties of tantalum and its alloys
[J].
Preparation of tantalum carbide films by reaction of electrolytic carbon coating with the tantalum substrate
[J].
Advances on surface treatment process of tantalum
[J].
金属钽表面处理工艺进展
[J].
(TaC/Ta2C) bilayer formed on carburized and annealed tantalum; development of a numerical growth model
[J].
A new strategy for preparing carbide coatings on easily oxidized tantalum by non-vacuum carburizing: Fe-coating-mediated carburization
[J].
Surface carburising of tantalum and its alloys
[J].
钽及其合金的表面渗碳强化
[J].
Surface treatment to improve the corrosion resistance of tantalum
[J].
提高钽耐蚀性的表面处理法
[J].
Surface modification of tantalum sheets by methane plasma carburizing
[J].
钽表面的甲烷等离子渗碳改性技术研究
[J].
Study of ionic carburisation modification of tantalum surfaces
[D].
钽表面离子渗碳改性研究
[D].
Vacuum carburization process of Ta and Ta-W alloys
[J].
Ta及Ta-W合金真空渗碳工艺研究
[J].
Microstructure and properties of carbide coatings on metal (Ta, Mo, Ti6Al4V) surface prepared by interstitial carburization
[D].
间隙原子渗碳法制备金属(Ta、Mo、Ti6Al4V)表面碳化层的组织和性能研究
[D].
Thermodynamic modeling of the Ta-Mo-C ternary system
[J].
A thermodynamic analysis of the Ta-W-C and the Ta-W-C-N systems
[J].
Critical thickness for ferroelectricity in perovskite ultrathin films
[J].
Novel electronic and magnetic properties of two-dimensional transition metal carbides and nitrides
[J].
High-entropy high-hardness metal carbides discovered by entropy descriptors
[J].
High-entropy materials have attracted considerable interest due to the combination of useful properties and promising applications. Predicting their formation remains the major hindrance to the discovery of new systems. Here we propose a descriptor-entropy forming ability-for addressing synthesizability from first principles. The formalism, based on the energy distribution spectrum of randomized calculations, captures the accessibility of equally-sampled states near the ground state and quantifies configurational disorder capable of stabilizing high-entropy homogeneous phases. The methodology is applied to disordered refractory 5-metal carbides-promising candidates for high-hardness applications. The descriptor correctly predicts the ease with which compositions can be experimentally synthesized as rock-salt high-entropy homogeneous phases, validating the ansatz, and in some cases, going beyond intuition. Several of these materials exhibit hardness up to 50% higher than rule of mixtures estimations. The entropy descriptor method has the potential to accelerate the search for high-entropy systems by rationally combining first principles with experimental synthesis and characterization.
Electron work function as an indicator for tuning the bulk modulus of MC carbide by metal-substitution: A first-principles computational study
[J].
Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set
[J].
From ultrasoft pseudopotentials to the projector augmented-wave method
[J].
Generalized gradient approximation made simple
[J].
Special points for Brillouin-zone integrations
[J].
Electronic and bonding analysis of hardness in pyrite-type transition-metal pernitrides
[J].
Structural, mechanical and electronic properties of (TaNbHfTiZr)C high entropy carbide under pressure: Ab initio investigation
[J].
Ab initio elastic tensor of cubic Ti0.5Al0.5N alloys: Dependence of elastic constants on size and shape of the supercell model and their convergence
[J].
Carbon stoichiometry and mechanical properties of high entropy carbides
[J].
Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles
[J].
First-principles study on the elastic and electronic properties of hexagonal ε-Fe3N
[J].
The elastic behaviour of a crystalline aggregate
[J].
Modeling hardness of polycrystalline materials and bulk metallic glasses
[J].
The alloy theoretic automated toolkit: A user guide
[J].
Special quasirandom structures
[J].
Bulk and surface electronic structure of hexagonal WC
[J].
Ab initio investigations of the phase stability in tantalum carbides
[J].
(W1 - x, Mx ) C carbides with desired combinations of compatible density and properties—A first-principles study
[J].
Elastic properties and plastic deformation of TiC- and VC-based pseudobinary alloys
[J].
Defect energetics and stacking fault formation in high-entropy carbide ceramics
[J].
The lattice distortion, mechanical and thermodynamic properties of A(Zr0.2Sn0.2Ti0.2Hf0.2Nb0.2)O3 (A = Sr, Ba) high-entropy perovskite with B-site disorder: First principles prediction
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}