


金属材料是现代工业和科技发展的重要基石,它在航天航空、轨道交通、国防军工等多个关键领域中发挥着核心作用。金属材料在服役过程中,通常会面临诸如温度、压力和应力场等多样的外场刺激,诱发金属材料的塑性变形或结构相变,继而导致材料性能的退化或失效[1]。因此准确地描述与理解金属材料在相变与形变过程中的微观物理机制,为设计出满足国家重大装备关键部件性能需求的材料提供理论指导,具有重要的战略意义。
近年来,虽然原位实验表征技术,如原位透射电子显微镜(in-situ transmission electron microscope)、同步辐射光源(synchrotron light source)、散裂中子源(spallation neutron source)等取得了显著进步,为实时监测和表征材料的结构提供了新途径,然而,揭示金属材料相变与形变的微观机制仍然面临着巨大的挑战[2~4]。这主要归因于2个关键因素:首先,时空间的限制——想要准确地捕获金属材料的相变与形变的微观机制,所需的空间-时间分辨率极高,例如,马氏体相变的速率和形核尺寸分别在声子时间尺度与纳米尺度[5]。其次,实验条件的精确控制存在挑战,特别是在极端服役条件下(如高温、高压等)对材料进行表征。
原子模拟(atomistic simulation),特别是分子动力学(molecular dynamics,MD)模拟技术,可以从飞秒和原子尺度观察物质在外场(温度、压力等)下的动力学响应,是“原位”观察材料微观结构状态的有效工具,为理解金属材料相变与形变的微观机制提供了新方法。目前常用的MD模拟技术主要为2大类:(1) 从头计算的分子动力学模拟(ab initio molecular dynamics simulation,AIMD),它通过近似求解薛定谔方程来描述材料的物理化学属性,被认为是一种最精确的模拟手段,可以定量描述材料的性能变化,但其非常昂贵,仅适用于短时间(几十皮秒)、小尺度(几百个原子)的动力学模拟[6,7];(2) 传统的分子动力学(CMD)模拟,它基于“半经验”或“经验”的简单物理函数描述原子间的相互作用,比如嵌入原子势(embedded atom method,EAM)和修正的嵌入原子势(MEAM),虽然可以实现高效率的大尺度模拟计算(几百纳米、几百纳秒),但是精度有限,特别是对金属材料的复杂相变和变形行为的准确描述仍存在一定的困难[8,9]。基于此,产生了分子动力学模拟技术中计算精度与效率的互制关系,限制了人们对于复杂动力学行为的原子尺度理解。
机器学习(machine learning,ML)赋能的原子模拟,特别是机器学习型分子力场(machine learning force fields,MLFFs)辅助的分子动力学模拟技术,作为一种新的模拟范式,打破了传统原子模拟技术在计算精度-效率上的互制关系[10,11],实现了从头计算精度(能量精度小于5 meV/atom,力精度小于1 eV/nm)的大尺度原子模拟[12,13]。继Behler和Parrinello[14]在MLFFs系统构造上的开创性工作之后,相关模拟方法已经在有机分子燃烧[15,16]、相变存储材料[17~19]、纳米催化[20,21]等重要科学领域的原子模拟中取得了显著成效,表明MLFFs有能力在材料模拟中推导范式转变,在小尺度模型和现实设备尺度模拟之间架起桥梁[22]。
本文综述了机器学习型分子力场强化的分子动力学模拟技术在金属材料相变与形变研究上的重要进展;简单介绍了机器学习型分子力场的基本原理与分类;重点阐述了该种方法在揭示金属材料相变机制与变形模式科学问题中的应用;讨论当前机器学习型分子力场的限制(和可能的陷阱),并概述未来在该领域的发展方向。
1 机器学习型分子力场技术
在过去的20年时间内,MLFFs作为一种变革性的原子模拟策略,完美地弥合了AIMD与CMD之间的鸿沟。MLFFs最早可以追溯到1992年,Ercolessi和Adams发现,利用大量的第一性原理计算数据集可以克服传统拟合方法在处理复杂解析函数时的挑战,获得与第一性原理计算精度相当的分子力场[23]。此后,Blank等[10]采用神经网络替代了传统的经验函数形式,奠定了MLFFs的基础框架。然而,真正推动MLFFs发展的是Behler和Parrinello[14]的开创性工作,他们基于高维中心对称函数拟合的神经网络势能面,为MLFFs的研究和应用开辟了新的道路。目前,国内外学者已经开发出多款机器学习势函数构建平台,如DeepMD[24]、GAP[25]、PiNN[26]、RuNNer[27]、MTP[28]、SchNet[29]、M3GNet[30]以及CHGNet[31]等。下文将探讨MLFFs的基本原理与要素,并以ML算法为导向,分别从核函数基与神经网络基2个方面介绍MLFFs技术。
1.1 MLFFs基本原理
MLFFs基本原理在于,无需依赖化学键特性或经验性假设,而是通过应用不同的机器学习算法,直接从第一性原理计算的数据集中学习原子局域环境与高维势能面之间的关联,即:
式中,E为模拟体系的总能量;N为模拟体系的原子数量;M为原子描述符的数量;αik 是i原子第k个描述符的线性组合系数,即机器学习模型获得的参数;Fik ( Ri )是原子局域描述符的函数, Ri 为原子i的位置。这种方法不仅避免了直接求解单电子近似的薛定谔方程,同时还能够实现与从头算方法相媲美的预测精度。
1.2 MLFFs基本要素
图1

图1
机器学习型分子力场(MLFFs)的构造示意图[32~34]
Fig.1
Schematic of machine-learning force fields (MLFFs)[32-34] (i, j, k represent three different atoms; rij denotes the distance between atom i and atom j; rik denotes the distance between atom i and atom k; θjik represents the angle formed by the central atom i with atoms k and j)
(1) 参考数据集。任何MLFFs的基本组成部分都是参考数据集。虽然MLFFs框架和其他技术细节对模型的潜在准确性负责,但是参考数据集及其质量定义了最终模型的可靠性和适用范围[35,36]。参考数据集通常来源于基于密度泛函理论(density functional theory,DFT)的高精度计算,其包含原子能量和受力信息等信息。目前参考数据集的收集在MLFFs构造过程仍是一个基本难题。首先是参考数据集来源于高精度的DFT计算,限制了大量数据的采集;此外,每一种材料的原子构型空间极其复杂,因此选择极具代表性的原子结构类似于大海捞针。为了解决这些难题,人们尝试性地设计了多种采样策略,其中包括基于AIMD的采样[37]、机器学习代理模型采样[38]、自适应采样[39]、元动力学采样[40]等[41~44]。更重要的是,在构造参考数据集时,人们需要根据关注的科学问题,对相应材料的相空间进行选择性采样,而非盲目性的随机采样[45]。
(2) 原子描述符。原子描述符在MLFFs构造中发挥着至关重要的作用。它将原子周围的局部和全局环境信息转换为数值特征,以便ML算法能够有效地处理[46,47]。此外,原子描述符需要满足旋转、平移和置换等不变性的物理约束[27,48]。原子描述符的质量直接影响着模型的性能,因此选择合适的原子描述符非常重要。理想的原子描述符应该能够充分反映原子间相互作用的复杂性,同时具有较低的计算成本[49]。目前存在多种不同的原子描述符,其中包括描述局部原子环境的中心原子对称函数(atom-centered symmetry functions,ACSFs)[48]和原子位置的平滑重叠(smooth overlap of atomistic positions,SOAP)[49]等;以及融合长程相互作用的Coulomb矩阵[50]和聚类扩展[51]等。近年来,基于网络生成的原子描述符,特别是图表示,因其固有的旋转、平移和置换不变性,正日益成为原子结构表示的流行选择。这种方法通过节点和边的信息来捕捉晶体结构的局部和整体特征,显示出了其高度的灵活性和有效性[52,53]。
1.3 核函数基的MLFFs
目前较为广泛使用的MLFFs回归模型是核函数基(kernel-based)方法。这种拟合方法需要精心设计原子描述符,因为它们在拟合过程中使用的“核技巧”,即是度量新结构的原子描述符与参考数据集原子描述符的相似程度[61]。在拟合过程中,使用简单的线性代数计算加权每一个核基函数的回归系数,其形式为:
式中,
1.4 基于神经网络的MLFFs
神经网络(neural network,NN)机器学习算法的提出,源于人类大脑的启发。它可以通过多层网络与非线性激活函数的叠加,即深度网络,高效地学习输入数据(原子描述符)和输出数据(能量与原子受力)之间的高度非线性关系。1995年,Blank等[10]首次尝试使用NN替代传统的经验解析表达式来拟合分子力场。此后,Lorenz等[65]进一步采用NN来拟合分子间的相互作用能量。然而,真正推动基于NN的MLFFs发展的是Behler与Parrinello[14]在2007年提出的,基于高维中心对称函数的NN势函数。这一开创性工作之后,大量基于NN的MLFFs相继被开发出来[26,27,58~60,66~69]。根据原子描述符构造的不同,这些MLFFs可被分类为基于原子描述符的NN-MLFFs和端对端的NN-MLFFs。
三体项表达式为[48]:
式中,
式中,rcut代表势函数的截断半径。当r > rcut时,fcut(r)的值为0。
图2

图2
DeepMD模型的示意图:下方的框架是其中深度神经网络的放大图;原子i所有相邻原子之间的距离矩阵{ Rij },即环境矩阵,首先转换为描述矩阵{ Dij },再传入到隐藏层计算原子能量Ei[66]
Fig.2
Schematic of the DeepMD model. The frame in the box is an enlargement of a deep neural network (DNN). The relative positions of all neighbors with respect to atom i, i.e., { Rij }, is first converted to descriptor matrix { Dij }, then passed to the hidden layers to compute Ei[66] (E—total energy)
图3

图3
基于图表示的原子描述符构造示意图。初始图由原子属性集V = {vi }、键属性集E = {(ek, rk, sk )}和全局状态属性 u 表示。在第一个更新步骤中更新键属性。信息从形成键的原子、状态属性和前一个键属性流向新的键属性。随后,通过这3个信息先后更新原子属性和全局状态属性。最终形成新的图[75]
Fig.3
Schematic of graph-based atomic descriptor. The initial graph is represented by the set of atomic attributes V = {vi }, bond attributes E = {(ek, rk, sk )}, and global state attributes u. In the first update step, the bond attributes are updated. Information flows from atoms that form the bond, the state attributes, and the previous bond attribute to the new bond attributes. Similarly, the second and third steps update the atomic and global state attributes, respectively, by information flow among all three attributes. The final result is a new graph representation[75] (vi is an atomic attribute vector for atom i in a system; ek is the bond attribute vector for bond k, rk and sk are the atom indices forming bond k; MEGNet—MatErials Graph Network)
1.5 不同MLFFs的效率与精度
势函数的效率与精度是决定分子动力学模拟尺度的重要因素。特别是在研究金属材料相变与形变行为时,捕捉微观缺陷的演化及其动力学机制通常需要较大的空间(约百纳米)和时间尺度(约纳秒)。因此,在计算效率与精度之间找到平衡显得尤为重要。基于此,Xie等[83]以金属W为例,比较了不同类型的MLFFs和几种常用的经验势函数(包括Lennard-Jones (LJ)、EAM和Morse势)之间的效率与精度关系(如图4[83]所示)。尽管MLFFs相对于传统的经验或半经验势函数具有更高的计算精度,但其计算效率降低了2~3个数量级。对于不同类型的MLFFs,尽管它们在计算精度上相差不大,但计算效率存在数量级的差异。这种差异源于不同MLFFs架构的基本差异。例如,GAP型的MLFFs,其计算复杂度为O(n³) (其中n是训练数据的数量),因此在大规模系统中的计算效率较低。
图4
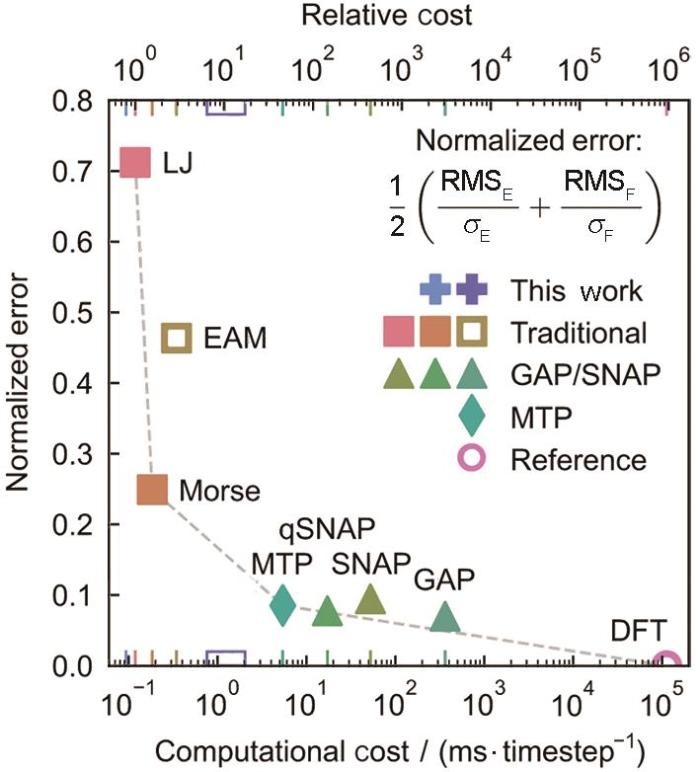
图4
不同MLFFs精度与效率的互制关系[83]
Fig.4
Trade-offs between accuracy and cost among different types of MLFFs. All potentials except EAM4 were refitted to the same tungsten data set. Computational costs were benchmarked with a 128-atom bcc-tungsten supercell[83] (EAM—embedded atom method; GAP—Gaussian approximation potential; SNAP, qSNAP—spectral neighbor analysis potential (SNAP) and its quadratic variant, respectively; MTP—moment tensor potential; DFT—density functional theory; LJ—Lennard-Jones function; RMSE—root-mean-squared error; RMSF—root-mean-square-fluctuation; σE—the range of the ground truth value of RMSE; σF—the range of the ground truth value of RMSF)
2 MLFFs技术在金属材料相变与形变中的应用
相变与形变是金属材料在服役过程中不可避免的物理过程,它们决定着材料的宏观力学性能与使用寿命。因此从原子尺度理解相关过程的物理机制,对设计和指导国家重大装备关键部件材料起着至关重要的作用。尽管原位实验观察是揭示金属材料与相变微观机制的重要手段,但目前对于相关机制的实验理解仍然有限,特别是在极端环境条件下(如高温、高压以及高应变速率等)。基于此,原子模拟技术的发展,特别是机器学习赋能的原子模拟技术,如MLFFs辅助的分子动力学模拟,为人们从原子尺度揭示金属材料相变与形变的微观物理图像带来曙光[84,85]。下文将面向金属材料,从形变到相变,重点阐述近几年机器学习势函数赋能的分子动力学模拟对理解相关机制做出的贡献。
2.1 金属材料的形变
金属形变是指在服役环境下,金属材料内部发生了不可逆塑性变形。根据加载速率的不同,金属材料的塑性变形可以分为准静态与冲击变形[86,87]。位错作为金属材料塑性变形过程中的主要载体,它的运动模式对金属材料的力学性能和断裂行为起着关键的作用[88,89]。人们利用MLFFs的分子动力学模拟对位错在不同载荷条件下(如准静态加载与动态冲击加载)的运动行为开展了大量研究。2018年,Maresca等[90]利用GAP研究了bcc Fe中单个螺位错在不同应力加载条件下的位移行为,成功复现了螺位错的紧凑核心(compact core)结构,如图5a和b[90]所示,并揭示了扭结对(kink-pair)的形核与增殖在螺位错的热激活运动模式中的关键作用。此外,Li等[91]采用GPR技术为NiAl高温合金开发了MLFFs,并研究了界面位错网络对镍基单晶高温合金应变速率敏感性的影响。结果表明,在低应力条件下,如图5c和d[91]所示,界面位错与基体位错之间存在弱的远程相互作用,而在高应力条件下,界面位错与传入位错发生强烈的相互作用,形成位错结。这种弱相互作用导致位错在界面附近堆积,而强烈的位错反应逐渐破坏了界面的连续性。
图5
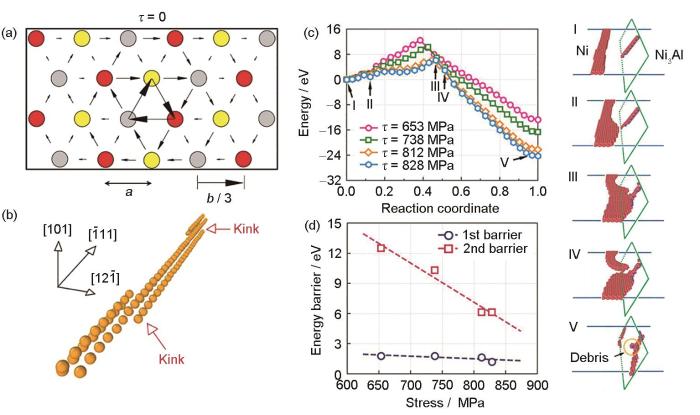
图5
MLFFs在传统金属材料准静态加载过程中位错动力学研究中的应用,包括:bcc-Fe的螺位错核心结构,bcc-Fe螺位错的扭结运动模式,1/2<110>螺位错与预先存在位错的Ni/Ni3Al半共格界面相互作用的应力与反应路径的依赖关系,及第一和第二相互作用阶段能量势垒随外部应力的变化[90,91]
Fig.5
Application of MLFFs in dislocation dynamics of conventional metallic materials under quasi-static loading condition
(a) screw dislocation core of bcc-Fe (τ—shear stress, a—Peierls valleys spacing, b—Burgers vector modulus)[90]
(b) atoms at the dislocation core during a simulation snapshot, evidencing dislocation glide by kink-pair mechanism[90]
(c) stress-dependent reaction path of the interaction between an incoming 1/2<110> screw dislocation and the semi-coherent Ni/Ni3Al interface with a preexisting interfacial dislocation[91]
(d) variation of energy barrier as a function of the external stress for the first and second interacting stages[91]
相比于传统的金属体系,多主元合金(multi-principal element alloys,MPEAs)和高熵合金(high-entropy alloys,HEAs)因其卓越的力学性能,在过去20年中成为研究热点。由于这些合金具有复杂的化学组成,传统的经验原子间势函数,如EAM和MEAM,面临着巨大挑战。因此,MLFFs成为了阐释其变形机制的强有力工具。例如Chen等[92]采用KRR方法拟合的MLFFs,研究了bcc-TiZrNb合金中位错环在应力作用下的行为。研究发现,晶格畸变的存在改变了刃位错的运动模式,导致了类似螺位错的钉扎-脱钉扎过程,这使得刃位错和螺位错的运动速率相当,这一发现颠覆了传统bcc金属中刃位错速率远大于螺位错的观念。虽然理想的MPEAs/HEAs被认为化学元素在三维空间中呈完全随机分布,但是真实条件下,并非每一种元素随机固溶,即化学短程有序(chemical short-range order,CSRO)通常存在于MPEAs/HEAs中,并且会影响缺陷的运动,继而影响力学性能。基于此,Yin等[93]利用矩张量势(moment tensor potential,MTP)[55]的形式开发了Nb-Ta-Mo-W高熵合金的MLFFs,并探究CSRO对位错行为的影响规律。如图6[93,94]所示,研究表明CSRO的存在会增强刃位错的运动速率,但降低螺位错的扭结形核率,继而降低螺位错的运动速率。此外,还观察到一种新型交叉滑移锁定机制,该机制可能为MoNbTaW高耐热高熵合金提供额外的强化效果。近期,Zheng等[95]同样利用MTP型的MLFFs对MoNbTi和TaNbTi高熵合金中位错的运动状态进行分析,从层错能的角度揭示了MoNbTi体系的应变强化效果比TaNbTi体系更加显著的原因,并发现随着短程有序现象增加,非稳态堆垛层错能的离散程度降低,继而减小加工硬化的程度。
图6

图6
MLFFs在高熵合金准静态加载过程中位错动力学研究中的应用[93,94]
Fig.6
Application of MLFFs in dislocation dynamics of high entropy alloys under qausi-static loading condition
(a) expansion of dislocation loop in Ni16.67Co16.67Fe36.67Ti30 high-entropy alloys (HEAs) under shear stress τxy = 600 MPa at T = 300 K[94] (t—the duration of the simulation)
(b) variation of velocity as a function of shear stress for both edge and screw dislocations at T = 300 K in Ni16.67Co16.67Fe36.67Ti30 HEAs[94]
(c) dislocation line with cross-slip locking viewed from the y and z directions[93] (d110—interplanar spacing of the 110 crystal plane)
(d, e) CSRO effects on screw dislocation velocity (d) and edge dislocation velocity (e) vs stress dependency at 1800 K (CSRO—chemical short-range order)[93]
除了位错,晶界也在金属材料的塑性变形中扮演着关键角色。Li等[96]采用谱相邻分析势(SNAP)对Ni-Mo合金的细晶强化机制进行了深入分析,并成功复现了实验中观察到的反Hall-Petch效应。研究揭示,当晶粒尺寸降至7 nm时,材料的变形机制由位错主导转变为晶界主导。此外,还发现通过溶质掺杂和退火处理合金,可以使元素在晶界偏聚,从而减轻晶界的内应力并增强其稳定性,这有助于降低屈服强度软化的临界尺寸。
除了位错,晶界滑移在金属材料塑性变形中也扮演着关键角色。Li等[97]采用谱相邻分析势(SNAP)对NbMoTaW多主元合金的纳米多晶力学行为进行了研究。研究发现(图7[97]),在Monte Carlo退火条件下,NbMoTaW合金的化学元素发生了重新分布,形成化学短程有序,即Nb与Mo元素偏聚到晶界,而W和Ta元素分布在晶内。这种化学短程有序可以缓解加载条件下von Mises应变的局域化,从而提高材料的屈服强度。在此基础上,研究人员对Ni-Mo合金的细晶强化机制进行了深入分析,成功复现了实验中观察到的反Hall-Petch关系,并揭示了当晶粒尺寸降至7 nm时,材料的变形机制由位错主导转变为晶界主导。此外,通过溶质掺杂和Monte Carlo退火处理合金,可以使元素在晶界偏聚,从而减轻晶界的内应力并增强其稳定性,这有助于降低屈服强度软化的临界尺寸。
图7

图7
MLFFs在金属纳米多晶力学行为中的应用[97]
Fig.7
Application of MLFFs in the mechanical behavior of nanocrystalline alloys[97]
(a) a polycrystalline model for the quaternary NbMoTaW multi-principal element alloys (MPEA) with atoms colored according to the common neighbor analysis algorithm to identify different structure types (cyan: bulk bcc; orange: grain boundary (GB))
(b) the same polycrystalline model after random initialization with equimolar quantities of Nb, Mo, W, and Ta. Atoms are colored by elements
(c) snapshot of polycrystalline model after hybrid Monte Carlo/molecular dynamics (MC/MD) simulations
相比于准静态加载,冲击加载下金属材料塑性变形机制的研究对分子力场提出了更高的要求。在这方面,MLFFs体现出了巨大优势。例如,Zhao等[98]采用核岭回归模型的机器学习势研究了bcc型高熵合金在冲击压缩下的位错行为。如图8a[98]所示,模拟发现与bcc型单质金属相比,冲击压缩下TiZrNb和NiCoFeTi高熵合金中存在异常的“扩展”刃位错结构(6~8 Burgers矢量),并且具有较高的稳定性。这种独特的位错核结构可以促进位错更快地运动,从而阻止了形变孪晶的早期形核。结合连续介质弹性理论,将这种“扩展”刃位错结构归因于高熵合金中纳米尺度化学不均匀性所引起的局部低弹性稳定性。进一步地,Li等[99]采用MLFFs探究了bcc ZrNb合金在冲击加载下变形模式的各向异性,以及化学元素分布对变形模式选择的影响。在沿<001>晶向的冲击加载下,主要观察到{111}<112>孪晶作为变形模式;而在沿<111>晶向的冲击加载中,于图8c[99]中可以发现,变形模式则包括了变形孪晶和相变,且变形模式的选择与Nb元素的分布有着密切的联系。这些发现为理解高熵合金在极端载荷条件下的变形行为提供了深入的洞见。
图8

图8
MLFFs在金属材料动态加载变形机制研究中的应用[98,99]
Fig.8
Application of MLFFs in dislocation dynamics of conventional metallic materials
(a) width of dislocation core as a function of the shock velocity in the elemental metals and bcc HEAs[98]
(b) phase diagram of dislocation core and spatial distribution of local shear modulus (Gatom)[98]
(c) microstructure of a Zr-10Nb (atomic fraction, %) single crystal at t = 12 ps in response to [001] β shock loading[99] (TB—twin boundary)
2.2 金属材料的相变
图9
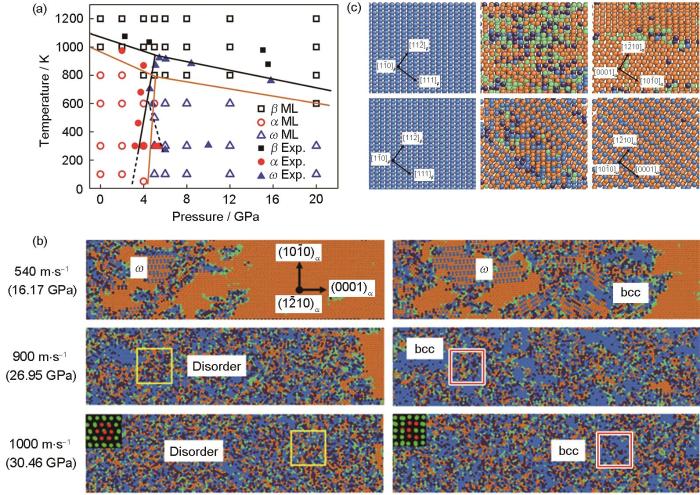
图9
MLFFs在过渡金属元素Zr相变机制研究中的应用[104,106]
Fig.9
Application of MLFFs in phase transition of zirconium
(a) predicted phase diagram of pure Zr as a function of pressure and temperature[106]
(b) snapshots of the phase transformation processes in [0001] α shocked Zr single crystals with different piston velocities[104] (Insets show the neiborhood information of the atoms before and after the transformation. They correspond to the hcp and bcc structures, respectively)
(c) typical microstructure evolution of β-Zr during cooling at pressure P = 0 GPa and P = 8.0 GPa using the present machine-learning (ML) potential[106]
随后,利用该MLFFs势函数研究Zr在冲击加载下的α→ω相变行为[105]。在低速冲击加载下(< 800 m/s),发现α→ω相转变的取向关系显示出弱的各向异性,并且在大多数情况下遵循Silcock关系(0001) α // (1
图10
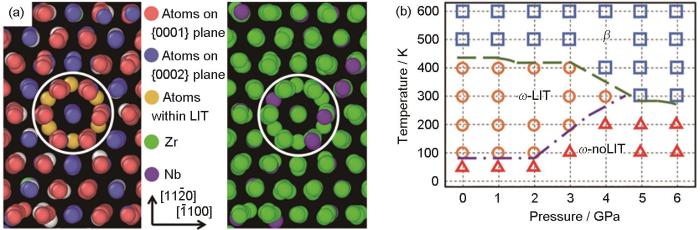
图10
MLFFs在过渡金属材料Zr-Nb合金相变机制研究中的应用:Zr-10Nb单晶形成局部层间扭转的分子动力学模拟及Zr-Nb合金的分子动力学模拟相图[108]
Fig.10
Application of MLFFs in phase transition of Zr-Nb alloy[108]
(a) MD simulation of the Zr-10Nb single crystal showing the formation of local interlayer twists (LITs)
(b) temperature-pressure phase diagram of Zr-Nb alloys from MD simulations
除了过渡金属Zr及其合金以外,MLFFs赋能的分子动力学模拟也推动了其他过渡金属材料,如Ti[109]、W[110]、U[111]、Ni-Ti[112]以及高熵合金[113]等的相变行为的理解。如Tang等[112]利用神经网络算法拟合了Ni-Ti形状记忆合金的MLFFs,并且预测了温度诱导、应力诱导和缺陷诱导的马氏体转变,与实验结果一致。Kostiuchenko等[113]使用机器学习低阶势对难熔bcc NbMoTaW合金的相变行为进行研究。通过考虑局部原子松弛的影响,发现B2有序相不是最稳定的状态,而是转变为一种新发现的、层状的半有序亚稳态。这个半有序相可以定性地描述为Nb-Mo-Ta-W和W-Ta-Mo-Nb叠层的重复,偶尔出现W-Ta-Mo-Ta-W和Nb-Mo-Ta-Mo-Nb叠层。
碱金属及碱土金属元素是主要的非过渡金属材料,并且在常压环境下被视为较简单的金属材料[114]。但是在压力作用下,它们通常会呈现出复杂的原子堆垛与电子结构转变,并表现出一些奇异的物理现象,如负斜率的熔化曲线(dT / d
图11

图11
MLFFs在碱金属K高压固态结构相变机制研究中的应用[117,118]
Fig.11
Application of MLFFs in phase transition of the high-pressure solid alkali metal potassium
(a) forcefield simulated phase diagram of potassium[118]
(b) top view and side view of the incommensurate host-guest (HG) structure K-III (a, b, c—lattice constants)
(c) correlation along the chains, showing long-range oscillatory order at 250 K (blue) and exponentially decaying short-ranged order at 750 K (red)[118](
(d, e) typical microstructure evolution of bicrystalline KIII-fcc upon isothermal annealing at 16 and 21 GPa[117] (Blue boxes outline the presence of disordered regions)
MLFFs辅助的MD模拟不仅可以揭示碱金属晶体结构间转变的机制,还助力于人们深入理解其液态结构的特性。Zong等[120]在碱金属K的液态区域内发现了压力诱发的原子液体-离子液体转变。如图12[120]所示,具体表现为加压过程中价电子结构的无序化和准局域态化,即部分价电子脱离原子实,随机填充并受限于原子间隙位置,形成赝负离子。该离子化过程能够将“电子团簇”以任意形状填充到间隙位置,实现更高效的离子堆垛,液体密度显著升高,进而导致熔点随着压力而降低(dT / dP < 0),形成高压“冷”液体。随后的同步辐射实验研究观察到了金属Na液体在“冷”液体稳定压力区间出现了折射率突降的现象,从实验上证实了“赝离子”液体的存在[121]。
图12

图12
MLFFs在碱金属K液态结构相变机制研究中的应用[120]
Fig.12
Application of MLFFs in phase transition of the liquid alkali metal potassium
(a) heat capacity (cp ) at different temperatures varies with pressure[120]
(b) normalized diffusion constant vs pressure for selected temperatures (D—diffusion constant) [120]
(c) radial distribution functions (RDFs) g(r) at selected pressures and 650 K
(d) heat map of distribution of coordination numbers vs pressure at 650 K[120]
(e) electron localization function (ELF) values at 2, 16, and 26 GPa are shown in the RGB scale from 0 (blue) to 0.70 (red), with 0.35 (green) [120]
3 MLFFs技术存在的壁垒与展望
计算效率:MLFFs通常需要处理大量的训练数据,其计算复杂度相比于传统的经验或半经验势函数往往较高。例如,Gauss过程回归的计算复杂度为O(n³),这在大规模系统中导致计算效率较低。此外,训练高精度的MLFFs需要大量的计算资源和时间,尤其是在处理复杂材料和多元体系时。随着硬件(如图形处理器(GPU)和张量处理器(TPU))的发展,尤其是并行计算和量子计算技术的进步,MLFFs的计算效率有望大幅提升。此外,分布式计算,即利用云计算和分布式计算平台,可以有效分担训练和计算的负担,从而提升整体效率。同时,MLFFs算法的优化,如稀疏Gaussian过程、神经网络压缩技术和高效的优化算法,以在保证精度的同时提高计算效率。
通用性:MLFFs的性能高度依赖于训练数据的质量和多样性,对于缺乏充分训练数据的新材料或新化学环境,模型可能表现不佳。此外,将MLFFs从一种材料体系应用到另一种材料体系可能需要重新训练,或者进行复杂的模型调整,限制了其通用性。因此,通过建立大规模、公开的高精度量子力学计算数据库,如Materials Project[122]和Open Quantum Materials Database (OQMD)[123]等[124~127],可以为MLFFs的训练提供丰富的数据支持,实现通用MLFFs的建立。另外,迁移学习和自适应模型也是一种可以实现MLFFs在不同材料体系之间具有更好通用性和适应性的策略。在此基础上,可以采用多种力场的耦合,即将MLFFs与传统经验力场相结合,利用各自的优势,提高模型的适用范围和准确性。
长程静电作用力的描述:传统MLFFs主要针对短程相互作用进行优化,处理长程静电相互作用(如van der Waals力和Coulomb力)时,计算复杂度和精度管理是一个挑战。准确描述长程静电作用力需要高精度的计算,这进一步增加了计算复杂度,影响整体效率。因此,开发混合模型,将MLFFs与长程力场模型结合,如结合Ewald求和[128]或Particle-Mesh Ewald (PME)方法[129],可以有效处理长程静电相互作用。此外,优化模型框架,探索和开发能够有效描述长程相互作用的MLFFs模型架构,如卷积神经网络(CNN)和图神经网络(GNN),使其在长程静电作用力描述上具有更高的精度和效率。
极端环境下MLFFs的可靠性:在近万Kelvins温度和百万个大气压下,材料的行为和相互作用变得极其复杂,传统的MLFFs可能难以准确描述这种条件下的原子结构变化。此外,目前的MLFFs通常仅基于原子局部结构进行表示,忽略了温度、压力等其他重要因素的影响。因此,可能需要引入更多变量作为输入特征,以实现对原子特征的完备描述。同时,在极高温和高压条件下,熵对体系的贡献显著增加,因此开发显性融合电子熵效应的MLFFs,实现近万Kelvins温度和百万个大气压下原子结构的准确描述是一个亟待深入发展的方向。
参考文献
In situ kinetic observations on crystal nucleation and growth
[J].
FMX—The frontier microfocusing macromolecular crystallography beamline at the national synchrotron light source II
[J].
Physical design and performance research of two counter-rotating T0 choppers for the multi-physics neutron diffractometer at China Spallation Neutron Source
[J].
The martensitic transition pathway in steel
[J].
The martensitic transformation (MT) lays the foundation for microstructure and performance tailoring of many engineering materials, especially steels, which are with > 1.8 billion tons produced per year the most important material class. The atomic-scale migration path is a long-term challenge for MT during quenching in high-carbon (nitrogen) steels. Here, we provide direct evidence of ($1\bar{1}2$) body-centred tetragonal (BCT) twinned martensite in carbon steels by transmission electron microscopy (TEM) investigation, and the increase in tetragonality with the C content matches X-ray diffraction (XRD) results. The specific {1$\bar{1}$2}BCT twin planes which are related to the elongated c axis provide essential structural details to revisit the migration path of the atoms in MT. Therefore, the face-centred cubic (FCC) to BCT twin to body-centred cubic (BCC) twin transition pathway and its underlying mechanisms are revealed through direct experimental observation and atomistic simulations. Our findings shed new light on the nature of the martensitic transition, thus providing new opportunities for the nanostructural control of metals and alloys.
Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients
[J].
Unified approach for molecular dynamics and density-functional theory
[J].
Modified embedded-atom potentials for cubic materials and impurities
[J].
Embedded-atom method: Derivation and application to impurities, surfaces, and other defects in metals
[J].
Neural network models of potential energy surfaces
[J].
Machine learning for molecular and materials science
[J].
Achieving DFT accuracy with a machine-learning interatomic potential: Thermomechanics and defects in bcc ferromagnetic iron
[J].
Machine learning predictions of molecular properties: Accurate many-body potentials and nonlocality in chemical space
[J].Simultaneously accurate and efficient prediction of molecular properties throughout chemical compound space is a critical ingredient toward rational compound design in chemical and pharmaceutical industries. Aiming toward this goal, we develop and apply a systematic hierarchy of efficient empirical methods to estimate atomization and total energies of molecules. These methods range from a simple sum over atoms, to addition of bond energies, to pairwise interatomic force fields, reaching to the more sophisticated machine learning approaches that are capable of describing collective interactions between many atoms or bonds. In the case of equilibrium molecular geometries, even simple pairwise force fields demonstrate prediction accuracy comparable to benchmark energies calculated using density functional theory with hybrid exchange-correlation functionals; however, accounting for the collective many-body interactions proves to be essential for approaching the “holy grail” of chemical accuracy of 1 kcal/mol for both equilibrium and out-of-equilibrium geometries. This remarkable accuracy is achieved by a vectorized representation of molecules (so-called Bag of Bonds model) that exhibits strong nonlocality in chemical space. In addition, the same representation allows us to predict accurate electronic properties of molecules, such as their polarizability and molecular frontier orbital energies.
Generalized neural-network representation of high-dimensional potential-energy surfaces
[J].
Complex reaction processes in combustion unraveled by neural network-based molecular dynamics simulation
[J].Combustion is a complex chemical system which involves thousands of chemical reactions and generates hundreds of molecular species and radicals during the process. In this work, a neural network-based molecular dynamics (MD) simulation is carried out to simulate the benchmark combustion of methane. During MD simulation, detailed reaction processes leading to the creation of specific molecular species including various intermediate radicals and the products are intimately revealed and characterized. Overall, a total of 798 different chemical reactions were recorded and some new chemical reaction pathways were discovered. We believe that the present work heralds the dawn of a new era in which neural network-based reactive MD simulation can be practically applied to simulating important complex reaction systems at ab initio level, which provides atomic-level understanding of chemical reaction processes as well as discovery of new reaction pathways at an unprecedented level of detail beyond what laboratory experiments could accomplish.
ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation
[J].
High-efficiency mechanical energy storage and retrieval using interfaces in nanowires
[J].By molecular dynamics simulations, we demonstrate a new concept for mechanical energy storage and retrieval using surface energy as reservoir in body-centered cubic (bcc) tungsten nanowire, achieving a combination of unique features such as large and constant actuation stress (>3 GPa), exceptionally large actuation strain (>30%) and energy density, and >98% energy storage efficiency. The underlying mechanism is a shear-dominant diffusionless transformation akin to martensitic transformation, but driven by surface rather than bulk free energies, and enabled by motion of coherent twin boundary, whose migration has been shown to possess ultralow friction in bcc metals. Aside from energy storage, such surface-energy driven displacive transformations are important for phase transformation and energy-matter control at the nanoscale.
Device-scale atomistic modelling of phase-change memory materials
[J].
Atomic insights into device-scale phase-change memory materials using machine learning potential
[J].
Understanding the composition and activity of electrocatalytic nanoalloys in aqueous solvents: A combination of DFT and accurate neural network potentials
[J].The shape, size, and composition of catalyst nanoparticles can have a significant influence on catalytic activity. Understanding such structure-reactivity relationships is crucial for the optimization of industrial catalysts and the design of novel catalysts with enhanced properties. In this letter, we employ a combination of first-principles computations and large-scale Monte-Carlo simulations with highly accurate neural network potentials to study the equilibrium surface structure and composition of bimetallic Au/Cu nanoparticles (NPs), which have recently been of interest as stable and efficient CO2 reduction catalysts. We demonstrate that the inclusion of explicit water molecules at a first-principles level of accuracy is necessary to predict experimentally observed trends in Au/Cu NP surface composition; in particular, we find that Au-coated core-shell NPs are thermodynamically favored in vacuum, independent of Au/Cu chemical potential and NP size, while NPs with mixed Au-Cu surfaces are preferred in aqueous solution. Furthermore, we show that both CO and O2 adsorption energies differ significantly for NPs with the equilibrium surface composition found in water and those with the equilibrium surface composition found in vacuum, suggesting large changes in CO2 reduction activity. Our results emphasize the importance of understanding and being able to predict the effects of catalytic environment on catalyst structure and activity. In addition, they demonstrate that first-principles-based neural network potentials provide a promising approach for accurately investigating the relationships between solvent, surface composition and morphology, surface electronic structure, and catalytic activity in systems composed of thousands of atoms.
Interpretable machine learning for knowledge generation in heterogeneous catalysis
[J].
Machine learning interatomic potential: Bridge the gap between small-scale models and realistic device-scale simulations
[J].
Interatomic potentials from first-principles calculations: The force-matching method
[J].
DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics
[J].
Gaussian approximation potentials: A brief tutorial introduction
[J].
PiNN: A python library for building atomic neural networks of molecules and materials
[J].Atomic neural networks (ANNs) constitute a class of machine learning methods for predicting potential energy surfaces and physicochemical properties of molecules and materials. Despite many successes, developing interpretable ANN architectures and implementing existing ones efficiently are still challenging. This calls for reliable, general-purpose, and open-source codes. Here, we present a python library named PiNN as a solution toward this goal. In PiNN, we designed a new interpretable and high-performing graph convolutional neural network variant, PiNet, as well as implemented the established Behler-Parrinello neural network. These implementations were tested using datasets of isolated small molecules, crystalline materials, liquid water, and an aqueous alkaline electrolyte. PiNN comes with a visualizer called PiNNBoard to extract chemical insight "learned" by ANNs. It provides analytical stress tensor calculations and interfaces to both the atomic simulation environment and a development version of the Amsterdam Modeling Suite. Moreover, PiNN is highly modularized, which makes it useful not only as a standalone package but also as a chain of tools to develop and to implement novel ANNs. The code is distributed under a permissive BSD license and is freely accessible at https://github.com/Teoroo-CMC/PiNN/ with full documentation and tutorials.
RuNNer—A program for constructing high-dimensional neural network potentials
[Z].
The MLIP package: Moment tensor potentials with MPI and active learning
[J].
SchNet—A deep learning architecture for molecules and materials
[J].
A universal graph deep learning interatomic potential for the periodic table
[J].Interatomic potentials (IAPs), which describe the potential energy surface of atoms, are a fundamental input for atomistic simulations. However, existing IAPs are either fitted to narrow chemistries or too inaccurate for general applications. Here we report a universal IAP for materials based on graph neural networks with three-body interactions (M3GNet). The M3GNet IAP was trained on the massive database of structural relaxations performed by the Materials Project over the past ten years and has broad applications in structural relaxation, dynamic simulations and property prediction of materials across diverse chemical spaces. About 1.8 million materials from a screening of 31 million hypothetical crystal structures were identified to be potentially stable against existing Materials Project crystals based on M3GNet energies. Of the top 2,000 materials with the lowest energies above the convex hull, 1,578 were verified to be stable using density functional theory calculations. These results demonstrate a machine learning-accelerated pathway to the discovery of synthesizable materials with exceptional properties.© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling
[J].
Quantum-mechanical exploration of the phase diagram of water
[J].The set of known stable phases of water may not be complete, and some of the phase boundaries between them are fuzzy. Starting from liquid water and a comprehensive set of 50 ice structures, we compute the phase diagram at three hybrid density-functional-theory levels of approximation, accounting for thermal and nuclear fluctuations as well as proton disorder. Such calculations are only made tractable because we combine machine-learning methods and advanced free-energy techniques. The computed phase diagram is in qualitative agreement with experiment, particularly at pressures ≲ 8000 bar, and the discrepancy in chemical potential is comparable with the subtle uncertainties introduced by proton disorder and the spread between the three hybrid functionals. None of the hypothetical ice phases considered is thermodynamically stable in our calculations, suggesting the completeness of the experimental water phase diagram in the region considered. Our work demonstrates the feasibility of predicting the phase diagram of a polymorphic system from first principles and provides a thermodynamic way of testing the limits of quantum-mechanical calculations.
AtomSets as a hierarchical transfer learning framework for small and large materials datasets
[J].
3D printed potential and free energy surfaces for teaching fundamental concepts in physical chemistry
[J].
USPEX—Evolutionary crystal structure prediction
[J].
Crystal structure prediction via particle-swarm optimization
[J].
Accelerated atomic data production in ab initio molecular dynamics with recurrent neural network for materials research
[J].
Metacontrol: A python based application for self-optimizing control using metamodels
[J].
DAS-PINNs: A deep adaptive sampling method for solving high-dimensional partial differential equations
[J].
Escaping free-energy minima
[J].We introduce a powerful method for exploring the properties of the multidimensional free energy surfaces (FESs) of complex many-body systems by means of coarse-grained non-Markovian dynamics in the space defined by a few collective coordinates. A characteristic feature of these dynamics is the presence of a history-dependent potential term that, in time, fills the minima in the FES, allowing the efficient exploration and accurate determination of the FES as a function of the collective coordinates. We demonstrate the usefulness of this approach in the case of the dissociation of a NaCl molecule in water and in the study of the conformational changes of a dialanine in solution.
Exploiting past visits or minimum-barrier knowledge to gain further boost in the temperature-accelerated dynamics method
[J].
Multicanonical ensemble: A new approach to simulate first-order phase transitions
[J].
Efficient, multiple-range random walk algorithm to calculate the density of states
[J].We present a new Monte Carlo algorithm that produces results of high accuracy with reduced simulational effort. Independent random walks are performed (concurrently or serially) in different, restricted ranges of energy, and the resultant density of states is modified continuously to produce locally flat histograms. This method permits us to directly access the free energy and entropy, is independent of temperature, and is efficient for the study of both 1st order and 2nd order phase transitions. It should also be useful for the study of complex systems with a rough energy landscape.
Nested sampling for general Bayesian computation
[J].
Accuracy and convergence of the Wang-Landau sampling algorithm
[J].
On representing chemical environments
[J].
Unified representation of molecules and crystals for machine learning
[J].
Atom-centered symmetry functions for constructing high-dimensional neural network potentials
[J].
Representations of materials for machine learning
[J].
Fast and accurate modeling of molecular atomization energies with machine learning
[J].
Generalized cluster description of multicomponent systems
[J].
Convolutional networks on graphs for learning molecular fingerprints
[A].
Graph neural networks for materials science and chemistry
[J].
Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials
[J].
Moment tensor potentials: A class of systematically improvable interatomic potentials
[J].
Learning scheme to predict atomic forces and accelerate materials simulations
[J].
Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons
[J].
Accurate and scalable graph neural network force field and molecular dynamics with direct force architecture
[J].
Directional message passing for molecular graphs
[DB/OL].
Fast and uncertainty-aware directional message passing for non-equilibrium molecules
[DB/OL].
Machine learning a general-purpose interatomic potential for silicon
[J].
Liquid-liquid phase transition in supercooled silicon
[J].Silicon in its liquid and amorphous forms occupies a unique position among amorphous materials. Obviously important in its own right, the amorphous form is structurally close to the group of 4-4, 3-5 and 2-6 amorphous semiconductors that have been found to have interesting pressure-induced semiconductor-to-metal phase transitions. On the other hand, its liquid form has much in common, thermodynamically, with water and other 'tetrahedral network' liquids that show density maxima. Proper study of the 'liquid-amorphous transition', documented for non-crystalline silicon by both experimental and computer simulation studies, may therefore also shed light on phase behaviour in these related materials. Here, we provide detailed and unambiguous simulation evidence that the transition in supercooled liquid silicon, in the Stillinger-Weber potential, is thermodynamically of first order and indeed occurs between two liquid states, as originally predicted by Aptekar. In addition we present evidence to support the relevance of spinodal divergences near such a transition, and the prediction that the transition marks a change in the liquid dynamic character from that of a fragile liquid to that of a strong liquid.
Microscopic mechanisms of pressure-induced amorphous-amorphous transitions and crystallisation in silicon
[J].Some low-coordination materials, including water, silica, and silicon, exhibit polyamorphism, having multiple amorphous forms. However, the microscopic mechanism and kinetic pathway of amorphous-amorphous transition (AAT) remain largely unknown. Here, we use a state-of-the-art machine-learning potential and local structural analysis to investigate the microscopic kinetics of AAT in silicon after a rapid pressure change. We find that the transition from low-density-amorphous (LDA) to high-density-amorphous (HDA) occurs through nucleation and growth, resulting in non-spherical interfaces that underscore the mechanical nature of AAT. In contrast, the reverse transition occurs through spinodal decomposition. Further pressurisation transforms LDA into very-high-density amorphous (VHDA), with HDA serving as an intermediate state. Notably, the final amorphous states are inherently unstable, transitioning into crystals. Our findings demonstrate that AAT and crystallisation are driven by joint thermodynamic and mechanical instabilities, assisted by preordering, occurring without diffusion. This unique mechanical and diffusion-less nature distinguishes AAT from liquid-liquid transitions.© 2024. The Author(s).
Representing high-dimensional potential-energy surfaces for reactions at surfaces by neural networks
[J].
Deep potential molecular dynamics: A scalable model with the accuracy of quantum mechanics
[J].
Interatomic potentials for ionic systems with density functional accuracy based on charge densities obtained by a neural network
[J].
Amp: A modular approach to machine learning in atomistic simulations
[J].
AENET-LAMMPS and AENET-TINKER: Interfaces for accurate and efficient molecular dynamics simulations with machine learning potentials
[J].
LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales
[J].
DeePMD-kit v2: A software package for deep potential models
[J].
86 PFLOPS deep potential molecular dynamics simulation of 100 million atoms with ab initio accuracy
[J].
Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning
[A]
Silicon liquid structure and crystal nucleation from ab initio deep metadynamics
[J].
Graph networks as a universal machine learning framework for molecules and crystals
[J].Graph networks are a new machine learning (ML) paradigm that supports both relational reasoning and combinatorial generalization. Here, we develop universal MatErials Graph Network (MEGNet) models for accurate property prediction in both molecules and crystals. We demonstrate that the MEGNet models outperform prior ML models such as the SchNet in 11 out of 13 properties of the QM9 molecule data set. Similarly, we show that MEGNet models trained on similar to 60 000 crystals in the Materials Project substantially outperform prior ML models in the prediction of the formation energies, band gaps, and elastic moduli of crystals, achieving better than density functional theory accuracy over a much larger data set. We present two new strategies to address data limitations common in materials science and chemistry. First, we demonstrate a physically intuitive approach to unify four separate molecular MEGNet models for the internal energy at 0 K and room temperature, enthalpy, and Gibbs free energy into a single free energy MEGNet model by incorporating the temperature, pressure, and entropy as global state inputs. Second, we show that the learned element embeddings in MEGNet models encode periodic chemical trends and can be transfer-learned from a property model trained on a larger data set (formation energies) to improve property models with smaller amounts of data (band gaps and elastic moduli).
Active learning across intermetallics to guide discovery of electrocatalysts for CO2 reduction and H2 evolution
[J].
Developing an improved crystal graph convolutional neural network framework for accelerated materials discovery
[J].
Perovskite synthesizability using graph neural networks
[J].
Transfer learning for materials informatics using crystal graph convolutional neural network
[J].
Open catalyst 2020 (OC20) dataset and community challenges
[J].
PotentialMind: Graph convolutional machine learning potential for Sb-Te binary compounds of multiple stoichiometries
[J].
GemNet: Universal directional graph neural networks for molecules
[A].
Ultra-fast interpretable machine-learning potentials
[J].
Phase transition study meets machine learning
[J].
Machine learning plastic deformation of crystals
[J].Plastic deformation of micron-scale crystalline solids exhibits stress-strain curves with significant sample-to-sample variations. It is a pertinent question if this variability is purely random or to some extent predictable. Here we show, by employing machine learning techniques such as regression neural networks and support vector machines that deformation predictability evolves with strain and crystal size. Using data from discrete dislocations dynamics simulations, the machine learning models are trained to infer the mapping from features of the pre-existing dislocation configuration to the stress-strain curves. The predictability vs strain relation is non-monotonic and exhibits a system size effect: larger systems are more predictable. Stochastic deformation avalanches give rise to fundamental limits of deformation predictability for intermediate strains. However, the large-strain deformation dynamics of the samples can be predicted surprisingly well.
Theoretical study of pressure effect on the dislocation core properties in semiconductors
[J].
Ab initio calculations of elastic constants of the bcc V-Nb system at high pressures
[J].
Dynamic transitions from smooth to rough to twinning in dislocation motion
[J].The motion of dislocations in response to stress dictates the mechanical behaviour of materials. However, it is not yet possible to directly observe dislocation motion experimentally at the atomic level. Here, we present the first observations of the long-hypothesized kink-pair mechanism in action using atomistic simulations of dislocation motion in iron. In a striking deviation from the classical picture, dislocation motion at high strain rates becomes rough, resulting in spontaneous self-pinning and production of large quantities of debris. Then, at still higher strain rates, the dislocation stops abruptly and emits a twin plate that immediately takes over as the dominant mode of plastic deformation. These observations challenge the applicability of the Peierls threshold concept to the three-dimensional motion of screw dislocations at high strain rates, and suggest a new interpretation of plastic strength and microstructure of shocked metals.
Energy radiation and limiting speeds of fast moving edge dislocations in tungsten
[J].
Screw dislocation structure and mobility in body centered cubic Fe predicted by a Gaussian Approximation Potential
[J].
Influence of interfacial dislocation network on strain-rate sensitivity in Ni-based single crystal superalloys
[J].
Correlating dislocation mobility with local lattice distortion in refractory multi-principal element alloys
[J].
Atomistic simulations of dislocation mobility in refractory high-entropy alloys and the effect of chemical short-range order
[J].Refractory high-entropy alloys (RHEAs) are designed for high elevated-temperature strength, with both edge and screw dislocations playing an important role for plastic deformation. However, they can also display a significant energetic driving force for chemical short-range ordering (SRO). Here, we investigate mechanisms underlying the mobilities of screw and edge dislocations in the body-centered cubic MoNbTaW RHEA over a wide temperature range using extensive molecular dynamics simulations based on a highly-accurate machine-learning interatomic potential. Further, we specifically evaluate how these mechanisms are affected by the presence of SRO. The mobility of edge dislocations is found to be enhanced by the presence of SRO, whereas the rate of double-kink nucleation in the motion of screw dislocations is reduced, although this influence of SRO appears to be attenuated at increasing temperature. Independent of the presence of SRO, a cross-slip locking mechanism is observed for the motion of screws, which provides for extra strengthening for refractory high-entropy alloy system.© 2021. The Author(s).
Unusual activated processes controlling dislocation motion in body-centered-cubic high-entropy alloys
[J].Atomistic simulations of dislocation mobility reveal that body-centered cubic (BCC) high-entropy alloys (HEAs) are distinctly different from traditional BCC metals. HEAs are concentrated solutions in which composition fluctuation is almost inevitable. The resultant inhomogeneities, while locally promoting kink nucleation on screw dislocations, trap them against propagation with an appreciable energy barrier, replacing kink nucleation as the rate-limiting mechanism. Edge dislocations encounter a similar activated process of nanoscale segment detrapping, with comparable activation barrier. As a result, the mobility of edge dislocations, and hence their contribution to strength, becomes comparable to screw dislocations.Copyright © 2020 the Author(s). Published by PNAS.
Multi-scale investigation of short-range order and dislocation glide in MoNbTi and TaNbTi multi-principal element alloys
[J].
Complex strengthening mechanisms in nanocrystalline Ni-Mo alloys revealed by a machine-learning interatomic potential
[J].
Complex strengthening mechanisms in the NbMoTaW multi-principal element alloy
[J].
Anomalous dislocation core structure in shock compressed bcc high-entropy alloys
[J].
The role of chemical disorder in β→ω phase transformation and deformation twinning in shock compressed Zr-Nb alloys
[Z].
From a single-band metal to a high-temperature superconductor via two thermal phase transitions
[J].
Quantum phase transition in a common metal
[J].
A first-principles theory of ferromagnetic phase transitions in metals
[J].
Crystal structures of titanium, zirconium, and hafnium at high pressures
[J].At high pressures, as determined by x-ray analysis, titanium and zirconium metal have a distorted, body-centered-cubic structure. This phase persists on pressure release. The normal hexagonal close-packed structures are recovered when the metals are heated. An electronic shift must occur in the transition. Hafnium metal showed no such transition.
Nucleation mechanism for hcp→bcc phase transformation in shock-compressed Zr
[J].
hcp→ω phase transition mechanisms in shocked zirconium: A machine learning based atomic simulation study
[J].
Developing an interatomic potential for martensitic phase transformations in zirconium by machine learning
[J].
Transformation pathway from alpha to omega and texture evolution in Zr via high-pressure torsion
[J].
Enhancing the stability of the ω phase of zirconium alloys via local interlayer twists
[J].
Machine learning models for predictive materials science from fundamental physics: An application to titanium and zirconium
[J].
Solid-solid phase transition of tungsten induced by high pressure: A molecular dynamics simulation
[J].
Phase diagram of uranium from ab initio calculations and machine learning
[J].
High accuracy neural network interatomic potential for NiTi shape memory alloy
[J].
Impact of lattice relaxations on phase transitions in a high-entropy alloy studied by machine-learning potentials
[J].
Structure of alkali metals in the liquid state
[J].
Commensurate-incommensurate phase transition of dense potassium simulated by machine-learned interatomic potential
[J].
Anomalous thermophysical properties and electride transition in fcc potassium
[J].
On the chain-melted phase of matter
[J].Various single elements form incommensurate crystal structures under pressure, where a zeolite-type "host" sublattice surrounds a "guest" sublattice comprising 1D chains of atoms. On "chain melting," diffraction peaks from the guest sublattice vanish, while those from the host remain. Diffusion of the guest atoms is expected to be confined to the channels in the host sublattice, which suggests 1D melting. Here, we present atomistic simulations of potassium to investigate this phenomenon and demonstrate that the chain-melted phase has no long-ranged order either along or between the chains. This 3D disorder provides the extensive entropy necessary to make the chain melt a true thermodynamic phase of matter, yet with the unique property that diffusion remains confined to 1D only. Calculations necessitated the development of an interatomic forcefield using machine learning, which we show fully reproduces potassium's phase diagram, including the chain-melted state and 14 known phase transitions.
Ab initio phase diagram and nucleation of gallium
[J].Elemental gallium possesses several intriguing properties, such as a low melting point, a density anomaly and an electronic structure in which covalent and metallic features coexist. In order to simulate this complex system, we construct an ab initio quality interaction potential by training a neural network on a set of density functional theory calculations performed on configurations generated in multithermal-multibaric simulations. Here we show that the relative equilibrium between liquid gallium, α-Ga, β-Ga, and Ga-II is well described. The resulting phase diagram is in agreement with the experimental findings. The local structure of liquid gallium and its nucleation into α-Ga and β-Ga are studied. We find that the formation of metastable β-Ga is kinetically favored over the thermodinamically stable α-Ga. Finally, we provide insight into the experimental observations of extreme undercooling of liquid Ga.
Free electron to electride transition in dense liquid potassium
[J].
Structural complexity in ramp-compressed sodium to 480 GPa
[J].The properties of all materials at one atmosphere of pressure are controlled by the configurations of their valence electrons. At extreme pressures, neighboring atoms approach so close that core-electron orbitals overlap, and theory predicts the emergence of unusual quantum behavior. We ramp-compress monovalent elemental sodium, a prototypical metal at ambient conditions, to nearly 500 GPa (5 million atmospheres). The 7-fold increase of density brings the interatomic distance to 1.74 Å well within the initial 2.03 Å of the Na ionic diameter, and squeezes the valence electrons into the interstitial voids suggesting the formation of an electride phase. The laser-driven compression results in pressure-driven melting and recrystallization in a billionth of a second. In situ x-ray diffraction reveals a series of unexpected phase transitions upon recrystallization, and optical reflectivity measurements show a precipitous decrease throughout the liquid and solid phases, where the liquid is predicted to have electronic localization. These data reveal the presence of a rich, temperature-driven polymorphism where core electron overlap is thought to stabilize the formation of peculiar electride states.© 2022. The Author(s).
Commentary: The materials project: A materials genome approach to accelerating materials innovation
[J].
The Open Quantum Materials Database (OQMD): Assessing the accuracy of DFT formation energies
[J].
A high-throughput infrastructure for density functional theory calculations
[J].
The computational materials repository
[J].
AFLOWLIB.ORG: A distributed materials properties repository from high-throughput ab initio calculations
[J].
ALKEMIE: An intelligent computational platform for accelerating materials discovery and design
[J].
Ewald summation for molecular simulations
[J].Ewald summation is an important technique for molecular simulation. In this article, expressions are provided for implementing Ewald summation for any inverse power potential in a range of different simulations. Energies, forces, stresses, and Hessian elements as well as truncation errors are considered. Focus is also given to methods for accelerating Ewald summation in Monte Carlo simulations, particularly in the grand canonical ensemble. Ewald techniques are applied to the simulation of CO2 adsorption and diffusion in the metal-organic framework, MOF-5. These simulations show that optimized Ewald summation can provide increased accuracy at similar computational cost compared to that of pair-based methods.
Staggered mesh Ewald: An extension of the smooth particle-mesh Ewald method adding great versatility
[J].We draw on an old technique for improving the accuracy of mesh-based field calculations to extend the popular Smooth Particle Mesh Ewald (SPME) algorithm as the Staggered Mesh Ewald (StME) algorithm. StME improves the accuracy of computed forces by up to 1.2 orders of magnitude and also reduces the drift in system momentum inherent in the SPME method by averaging the results of two separate reciprocal space calculations. StME can use charge mesh spacings roughly 1.5× larger than SPME to obtain comparable levels of accuracy; the one mesh in an SPME calculation can therefore be replaced with two separate meshes, each less than one third of the original size. Coarsening the charge mesh can be balanced with reductions in the direct space cutoff to optimize performance: the efficiency of StME rivals or exceeds that of SPME calculations with similarly optimized parameters. StME may also offer advantages for parallel molecular dynamics simulations because it permits the use of coarser meshes without requiring higher orders of charge interpolation and also because the two reciprocal space calculations can be run independently if that is most suitable for the machine architecture. We are planning other improvements to the standard SPME algorithm, and anticipate that StME will work synergistically will all of them to dramatically improve the efficiency and parallel scaling of molecular simulations.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}