


晶界对材料的力学行为和塑性变形机制具有显著的影响。当晶粒尺寸大于100 nm时,晶界阻碍位错运动,这导致位错在界面处塞积从而起到强化作用。当晶粒尺寸在数十纳米时,晶界对位错滑移的阻碍减弱,变形机制从位错塞积转变为非相关位错滑移,即Orowan-type或Whisker-type机制[26]。随着晶粒尺寸继续降低至10 nm以下时,塑性变形由晶界迁移和晶粒转动主导,这导致反Hall-Petch关系出现[27]。对纳米晶材料而言,晶界占有较高的体积分数。例如,当晶粒尺寸为10 nm时,晶界处的原子数占总原子的14%~27%。在外加载荷作用下晶界可以发射或吸收位错,且晶界的滑移也会释放应力,从而影响材料的力学性能[28]。然而,高熵合金较高的结构熵和晶格畸变导致其晶界的结构和力学行为与传统金属材料存在较大差异[11]。因此,有必要研究纳米晶高熵合金中晶界对宏观力学性能和微观变形机制的影响。
在纳米尺度下,分子动力学模拟可对各种变形系统进行准确、高效地追踪[14,25,29~32]。借助这一模拟方法,Yuasa等[29,30]对Cu/Cu双晶和Co/Cu双晶晶界处的位错发射和晶界滑移行为进行了系统研究,在Co/Cu双晶中,位错由界面处的层错产生。与Cu/Cu双晶相比,Co/Cu双晶中层错的形成并未促进位错发射,且晶界的滑动和迁移行为与自由体积的关系较小。Tan等[31]研究了熵值对FeCuCoCrNi高熵合金中晶界的结构和迁移机制的影响。随着熵值增加,晶界发生粗化,且晶界的迁移机制由集体迁移转变为分散迁移。Lee等[32]模拟了沿<110>和<111>方向单轴拉伸时具有不同对称扭转晶界的FeMnCoCrNi高熵双晶的微观力学行为,结果表明,Schokley不全位错优先在晶界处形核,然后向晶内滑移并留下层错,且位错形核所需的临界剪切应力随着双晶晶界角度的不同而发生变化。
为了揭示晶界对不同取向的高熵合金双晶塑性变形的影响,本工作借助分子动力学方法对具有不同初始取向组合的等原子比FeMnCoCrNi双晶微柱进行了拉伸变形模拟。模拟结果将阐释以下3个问题:(1) 不同取向的高熵双晶如何协调变形;(2) 双晶模型中晶界与变形方向的位向关系(平行或垂直)对材料的宏观性能和微观变形的影响;(3) 具有不同层错能和晶格畸变的高、低熵双晶材料塑性变形机制的差异。以上研究结果将有助于深入理解晶界对高熵合金塑性变形的影响,从而为设计高强韧性的高熵合金材料提供指导。
1 模型与方法
对等主元FeMnCoCrNi高熵合金的拉伸模拟结果表明,[001]和[123]取向单晶微柱的主要变形机制分别为层错的开动和ε-马氏体相变。且与其他取向相比,[110]取向单晶中的孪生行为最为显著,而[111]取向单晶中位错开动的临界应力最大[25]。因此,本工作选取[001]、[110]、[111]和[123] 4种单晶取向进行两两组合来构建双晶模型,即模拟涉及[001] + [110]、[001] + [111]、[001] + [123]、[111] + [110]、[123] + [110]和[111] + [123] 6种取向组合。为了研究晶界与加载方向的位向关系对材料变形的影响,在固定单轴拉伸方向的前提下改变相邻晶粒的相对位置,如图1所示,即分别为拉伸方向与晶界垂直和拉伸方向与晶界平行。图中A和B分别代表具有不同初始取向的单晶,尺寸均为15 nm (X) × 15 nm (Y) × 15 nm (Z)。值得注意的是,改变不同取向晶粒的相对位置后,晶界的粗糙程度虽然存在细微差异,但晶界的宽度均仅为2~3层原子间距,且不同双晶模型在变形前均未发现晶界附近有明显的位错,因此忽略了晶界微观结构的细微差异对大变形量下微观变形机制的影响。
图1
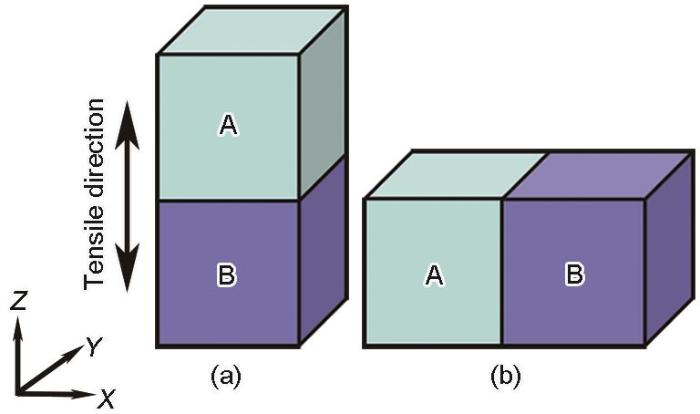
图1
单轴拉伸载荷下的双晶微柱模型示意图
Fig.1
Schematics of the bicrystal micropillar models deformed under uniaxial tension (A and B represent the single crystals with different initial orientations and sizes of 15 nm (X) × 15 nm (Y) × 15 nm (Z))
(a) grain boundary is perpendicular to the deformation direction
(b) grain boundary is parallel to the deformation direction
为研究晶粒尺寸对变形系统演化的影响,选取取向为[110],尺寸分别为5 nm (X) × 5 nm (Y) × 10 nm (Z)和15 nm (X) × 15 nm (Y) × 30 nm (Z)的FeMnCoCrNi单晶进行单轴拉伸变形的模拟对比。
为了揭示层错能和晶格畸变对双晶模型变形机制的影响,选取具有相同几何尺寸和拉伸方向的等原子比FeCuCoCrNi高熵合金和纯Cu进行了对比研究。采用MEAM (modified embedded atom method)势描述FeMnCoCrNi和Cu模型内原子的相互作用[33,34]。对FeCuCoCrNi高熵合金则选取EAM (embedded atom method)势[23],基于此势函数FeCuCoCrNi的平均点阵位移约为0.0027 nm,即晶格畸变程度约为0.7%,与采用第一性原理计算得到的FeMnCoCrNi晶格畸变程度相近[23]。上述势函数被广泛用来描述合金的力学行为和塑性变形机制。经计算,FeMnCoCrNi、FeCuCoCrNi和纯Cu的晶格常数依次为0.3595、0.3552和0.3615 nm,层错能分别为-45、62和41 mJ/m2,这些模拟结果与文献[23,33~35]报道一致。
分子动力学计算采用LAMMPS (large-scale atomic/molecular massively parallel simulator)软件包[36]模拟实现。对所有模型均进行300 K下的单轴拉伸变形模拟,变形方向(Z)为周期性边界条件,其他2个正交方向(X、Y)为自由边界条件。为了获得大塑性变形下不同变形系统的演化及其对力学性能的影响,施加的最大应变(工程应变ε)为100%。变形前,先对模型进行能量最小化处理,然后在300 K下进行模拟退火处理,以消除不合理的原子配置并释放内应力,使Z方向上的压力为0。最后对模型进行单轴拉伸模拟,模拟时间步长为5 fs。选取的应变速率为108 s-1。模拟结果的可视化通过OVITO (open visualization tool)实现[37],不同种类的原子结构和位错类型分别采用CNA (common neighbor analysis)[38]和DXA (dislocation analysis)[39]算法进行分析。
2 结果与讨论
2.1 不同晶粒尺寸单晶的力学行为
图2

图2
[110]取向下不同晶粒尺寸FeMnCoCrNi单晶的应力-应变曲线和相应不同形变系统的体积分数随应变的演化
Fig.2
Stress-strain curves (a, c) and corresponding volume fraction evolutions (b, d) of the [110]-oriented FeMnCoCrNi single crystals with domain sizes of 5 nm (X) × 5 nm (Y) × 10 nm (Z) (a, b) and 15 nm (X) × 15 nm (Y) × 30 nm (Z) (c, d) (SF—stacking fault, TB—twin boundary)
2.2 晶界垂直于拉伸方向时双晶模型的力学行为
当晶界垂直于拉伸变形方向时,不同FeMnCoCrNi双晶模型的应力-应变曲线和弹塑性转变点处的应力分别如图3和表1所示。在弹性变形阶段,随着应变增加,各模型的应力均近似呈线性增大直至达到弹塑性转变点。之后,应力便呈现出不同程度的下降,模型进入塑性变形阶段(图3)。在弹塑性转变点处,各模型的应力从大到小依次为[001] + [111]、[111] + [123]、[001] + [123]、[123] + [110]、[001] + [110]和[111] + [110]双晶。此时,双晶模型的应力均小于其组成单晶的应力(表1)。在塑性变形阶段,随着载荷增大,除[001] + [111]模型外,其余各模型呈现出不同程度的加工硬化,如图3中阴影区域所示,这归因于模型内形成了层错、孪晶和ε-马氏体等多种变形系统。由于[001] + [111]模型中较早发生了颈缩,因此其应力随应变的增加呈逐渐下降的趋势。当工程应变达到45%后,各模型的应力均随应变的增加而快速下降,直至断裂。值得注意的是,在应变为2%~5%时,[111] + [110]模型的应力呈现急剧下降的趋势,随后在应变为5%~20%时呈现出最为显著的加工硬化(图3)。
图3
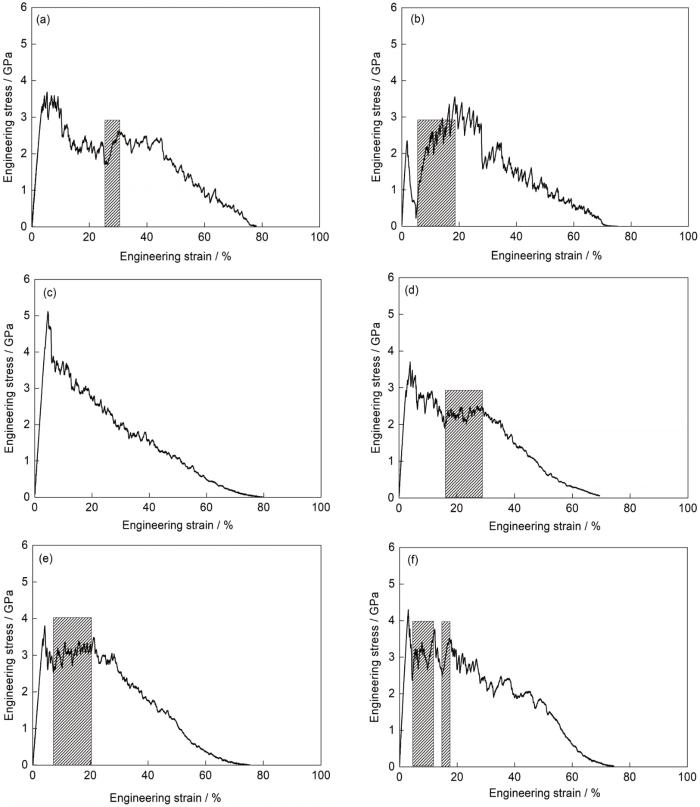
图3
晶界与拉伸方向垂直时不同取向组合双晶的应力-应变曲线
Fig.3
Stress-strain curves of the bicrystals with different orientation combinations when the grain boundary is perpendicular to the tensile direction (The shaded areas indicating work hardening)
(a) [001] + [110] (b) [111] + [110]
(c) [001] + [111] (d) [123] + [110]
(e) [001] + [123] (f) [111] + [123]
表1 晶界与拉伸方向垂直时单晶与双晶在弹塑性转变点处的应力
Table 1
| Crystal type | Orientation | Stress / GPa |
|---|---|---|
| Single crystal | [001] | 5.13 |
| [110] | 4.11 | |
| [111] | 7.43 | |
| [123] | 4.43 | |
| Bicrystal | [001] + [110] | 3.59 |
| [001] + [111] | 5.12 | |
| [001] + [123] | 3.80 | |
| [111] + [110] | 2.33 | |
| [123] + [110] | 3.70 | |
| [111] + [123] | 4.30 |
为了揭示单轴拉伸过程中微观变形系统的演化,各模型在弹塑性转变点处的原子结构和位错线分布分别如图4a~f和a1~f1所示。在所有模型中,Schockley不全位错优先在晶界处形核并随着变形量增大向晶粒内部滑移,且位错滑移后形成层错。由于晶体结构的差异,不同相之间的界面处通常存在失配位错结构[29]。由于FeMnCoCrNi双晶晶界两侧晶粒的取向不同,因此晶界处存在失配应变,进而导致位错在外加载荷作用下更易形核并向晶内滑移,即晶界处位错的形核由失配应变和变形共同导致。位错的滑移面和滑移方向与由Schmidt定律计算得到的优先开动的滑移系统一致。在[001] + [110]、[001] + [111]、[001] + [123]和[123] + [110]双晶模型中仅有少量位错在自由表面处形核(图4a1、b1、c1和e1)。对单晶模型的模拟结果表明,位错优先在自由表面形核[25,33]。与单晶相比,双晶模型中位错形核的临界应力较小,即晶界处更易促进位错形核。弹塑性转变点处[001] + [110]、[001] + [111]、[001] + [123]、[111] + [110]、[123] + [110]和[111] + [123]双晶的位错密度分别为1.003 × 1016、3.132 × 1016、0.975 × 1016、8.722 × 1016、0.997 × 1016和5.239 × 1016 m-2。与其他模型相比,[111] + [110]模型晶界处的位错密度最高。随着载荷增加,此模型中被激发出大量的位错且位错滑移方向指向[111]晶粒(图4d1)。这导致材料的应力迅速下降(图3)。当应变增加至约20%时,[111]和[110]晶粒内形成的层错、ε-马氏体和孪晶(图5)共同导致此双晶模型呈现较为显著的加工硬化。
图4

图4
晶界与拉伸方向垂直时不同取向组合的FeMnCoCrNi双晶在弹塑性转变点处的原子结构和位错线分布
Fig.4
Atomic structures (a-f) and corresponding dislocation line distributions (a1-f1) for the FeMnCoCrNi bicrystals with different orientation combinations at elastic-to-plastic transition point when the grain boundary is perpendicular to the tensile direction (a, a1) [001] + [110], engineering strain ε = 4.4% (b, b1) [001] + [111], ε = 4.6% (c, c1) [001] + [123], ε = 4.2% (d, d1) [111] + [110], ε = 1.9% (e, e1) [123] + [110], ε = 3.9% (f, f1) [111] + [123], ε = 3.0%
图5
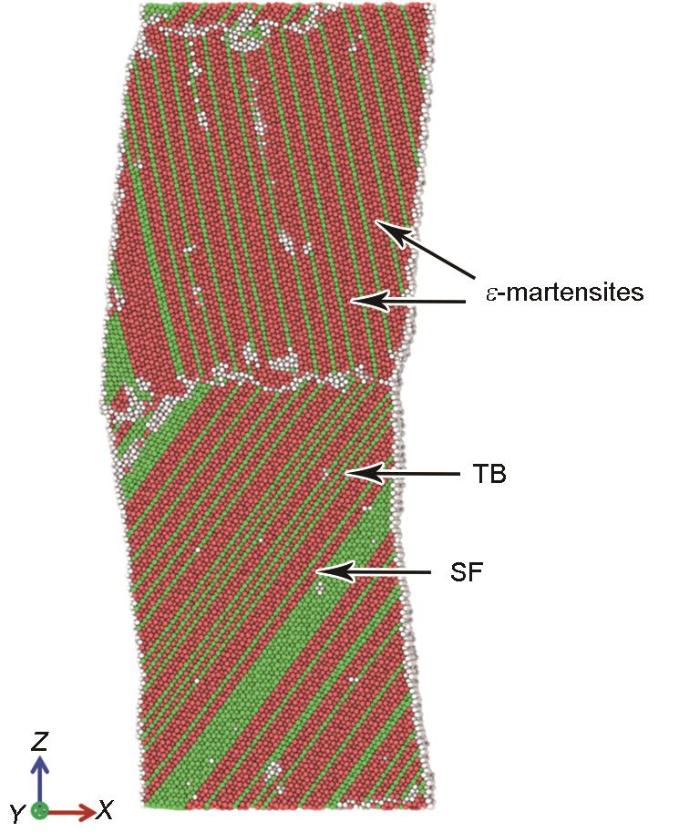
图5
应变为20%时[111] + [110]双晶模型的原子结构
Fig.5
Atomic structure for the [111] + [110] bicrystal at ε = 20%
当外加应变达到45%时,各模型的原子结构和位错线分布分别如图6a~f和a1~f1所示。所有模型均呈现出不同程度的颈缩,同时晶粒内部的变形不均匀。此外,晶界还呈现出宽化和弯曲的特征。除[001] + [111]和[111] + [110]双晶模型的颈缩发生在非晶界处外,其余模型的颈缩均在晶界发生,且颈缩位置的部分原子呈现非晶态,这表明当晶界与变形方向垂直时,颈缩更倾向于在晶界发生。双晶模型中各晶粒变形系统的开动与相同取向单晶的模拟结果[25]一致。其中,与[001]取向的晶粒相比,[110]、[111]和[123]取向的晶粒中开动了较多的ε-马氏体,且在这4种不同取向的晶粒中[110]晶粒内的位错密度最低。在[111] + [110]模型中,[111]晶粒内的位错密度与其他双晶模型中该取向晶粒相比较低(图6d1),这归因于[111]取向的晶粒内开动了较多的层错和ε-马氏体(图6d)。由此可见,[111] + [110]双晶模型中的晶界促进了[111]晶粒中层错和马氏体相变的发生,进而抑制了位错滑移。
图6

图6
晶界与拉伸方向垂直时不同取向组合的FeMnCoCrNi双晶在45%工程应变时的原子结构和位错线分布
Fig.6
Atomic structures (a-f) and corresponding dislocation line distributions (a1-f1) for the FeMnCoCrNi bicrystals with different orientation combinations at ε = 45% when the grain boundary is perpendicular to the tensile direction (a, a1) [001] + [110] (b, b1) [001] + [111] (c, c1) [001] + [123] (d, d1) [111] + [110] (e, e1) [123] + [110] (f, f1) [111] + [123]
2.3 晶界平行于拉伸方向时双晶模型的力学行为
当双晶模型的晶界与变形方向平行时,单轴拉伸载荷下各模型的应力-应变曲线和弹塑性转变点处的应力分别如图7和表2所示。在弹性变形阶段,模型的应力随应变的增加呈线性上升的趋势(图7)。当应力达到弹塑性转变点时,各模型的应力按由高到低排列依次为[001] + [111]、[111] + [123]、[111] + [110]、[001] + [123]、[123] + [110]和[001] + [110] (表2)。进入塑性变形阶段后,随着应变增加,由于[111] + [110]模型中形成了大量的ε-马氏体,因此呈现出最为显著的加工硬化,这将在下文详细描述。其次为[123] + [110]和[001] + [110]模型,如图7中阴影区域所示,其他模型的应力随变形增加总体呈现下降的趋势。当应变达到100%时,所有模型的应力仍保持在1 GPa以上。与晶界垂直于应力轴的模拟相比,晶界平行于变形方向时模型在弹塑性转变点处的应力水平略高,且在塑性变形阶段的流变应力随变形的演化更加平稳,这归因于模拟中晶界的位向和模型几何形状的差异。
图7
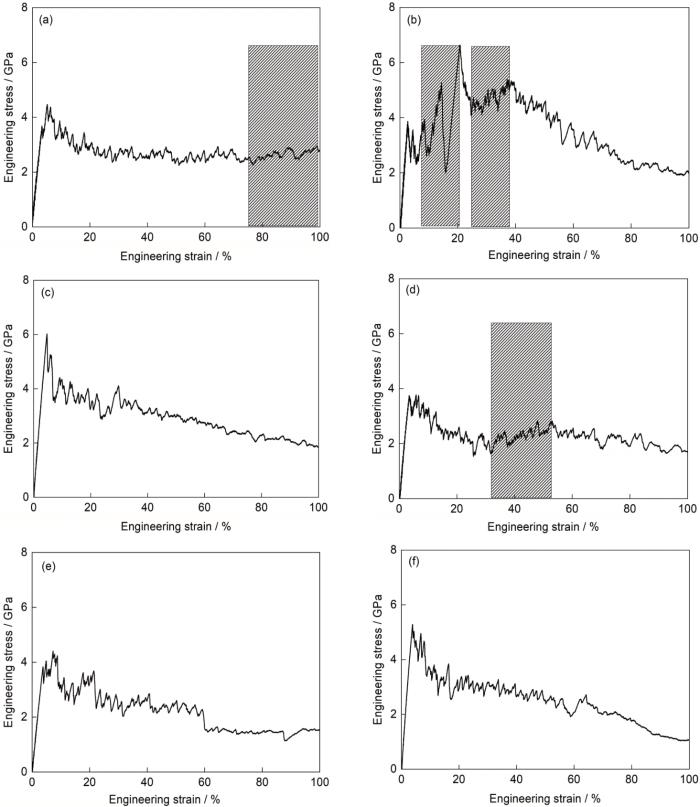
图7
当晶界与拉伸方向平行时不同取向组合双晶的应力-应变曲线
Fig.7
Stress-strain curves of the bicrystals with different orientation combinations when the grain boundary is parallel to the tensile direction (The shaded areas indicating work hardening)
(a) [001] + [110] (b) [111] + [110]
(c) [001] + [111] (d) [123] + [110]
(e) [001] + [123] (f) [111] + [123]
表2 当晶界与拉伸方向平行时单晶与双晶在弹塑性转变点处的应力
Table 2
| Crystal type | Orientation | Stress / GPa |
|---|---|---|
| Single crystal | [001] | 5.13 |
| [110] | 4.11 | |
| [111] | 7.43 | |
| [123] | 4.43 | |
| Bicrystal | [001] + [110] | 3.62 |
| [001] + [111] | 6.03 | |
| [001] + [123] | 3.83 | |
| [111] + [110] | 3.87 | |
| [123] + [110] | 3.72 | |
| [111] + [123] | 5.28 |
当外加载荷达到弹塑性转变点时,位错开始形核,如图8所示,此时,晶界仍为位错优先形核的位置(图8a1~f1),此外,在[001] + [110]、[001] + [111]、[001] + [123]和[111] + [123]双晶模型中也可观察到少量位错在自由表面处形核(图8a1~c1、f1)。与晶界垂直于拉伸方向的情形(图4)相似,位错滑移后会在晶粒内留下层错。在双晶模型中,位错优先向易于开动位错的晶粒内部滑移(图8a~f)。
图8
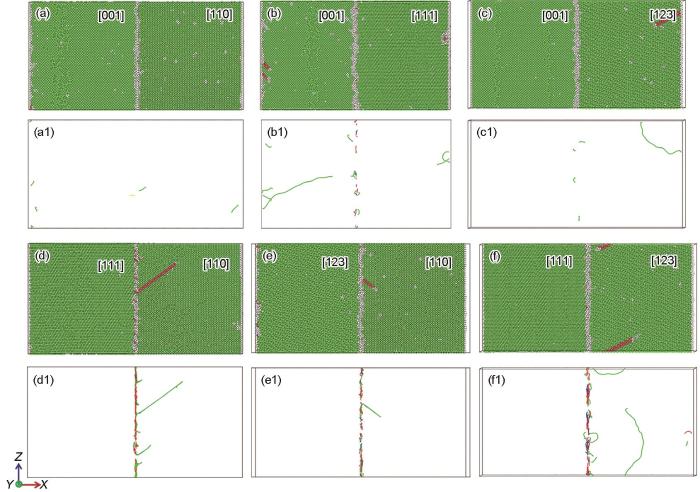
图8
晶界与拉伸方向平行时不同取向组合的FeMnCoCrNi双晶在弹塑性转变点处的原子结构和位错线分布
Fig.8
Atomic structures (a-f) and corresponding dislocation line distributions (a1-f1) for the FeMnCoCrNi bicrystals with different orientation combinations at elastic-to-plastic transition point when the grain boundary is parallel to the tensile direction
(a, a1) [001] + [110], ε = 3.3% (b, b1) [001] + [111], ε = 4.6% (c, c1) [001] + [123], ε = 3.8%
(d, d1) [111] + [110], ε = 2.7% (e, e1) [123] + [110], ε = 3.5% (f, f1) [111] + [123], ε = 3.9%
图9

图9
晶界与拉伸方向平行时不同取向组合的FeMnCoCrNi双晶在100%工程应变时的原子结构和位错线分布
Fig.9
Atomic structures (a-f) and corresponding dislocation line distributions (a1-f1) for the FeMnCoCrNi bicrystals with different orientation combinations at ε = 100% when the grain boundary is parallel to the tensile direction (a, a1) [001] + [110] (b, b1) [001] + [111] (c, c1) [001] + [123] (d, d1) [111] + [110] (e, e1) [123] + [110] (f, f1) [111] + [123]
综上所述,当晶界与变形方向的位向关系不同时,虽然在弹塑性转变点处,位错均更易在晶界处形核,但在塑性变形过程中晶界对双晶模型的力学行为和塑性变形机制的影响存在显著差异。具体而言,当晶界垂直于变形方向时,在拉应力作用下,晶界处更易积累缺陷,从而发生颈缩,导致双晶模型的应力下降。而当晶界平行于变形方向时,拉应力与晶界平行,垂直于晶界方向的受力较小,在变形过程中材料出现了晶界宽化和弯曲,进而协调了双晶体的塑性变形。这表明通过调控合金中晶粒的取向和晶界与变形方向的位向关系可提升材料的塑性变形能力。
2.4 层错能和晶格畸变对双晶变形机制的影响
变形前,3种材料不同双晶模型界面处的原子结构和原子势能分布如图10所示。当晶界与变形方向垂直时,FeMnCoCrNi和FeCuCoCrNi 2种双晶模型的晶界与Cu相比较为粗糙,同时,FeMnCoCrNi的晶界比FeCuCoCrNi的粗糙。此外,与[123] + [110]双晶模型相比,3种金属的[111] + [110]模型中晶界处的原子结构较为平直和光滑(图10a)。晶界处的原子势能分布表明不同材料双晶模型的晶界能存在差异。如图10b所示,与FeMnCoCrNi和FeCuCoCrNi高熵合金相比,Cu中晶界处的原子势能较高,且晶界的原子结构对原子势能无显著影响。当晶界与变形方向平行时,不同双晶模型晶界处的原子结构(图10c)粗糙程度的差异减小,而原子势能分布(图10d)的规律与晶界垂直于变形方向时一致。这表明,金属材料内晶界的原子结构不仅与熵值有关,还受元素种类及晶界两侧晶体取向的影响,而原子势能的分布主要取决于元素种类。
图10
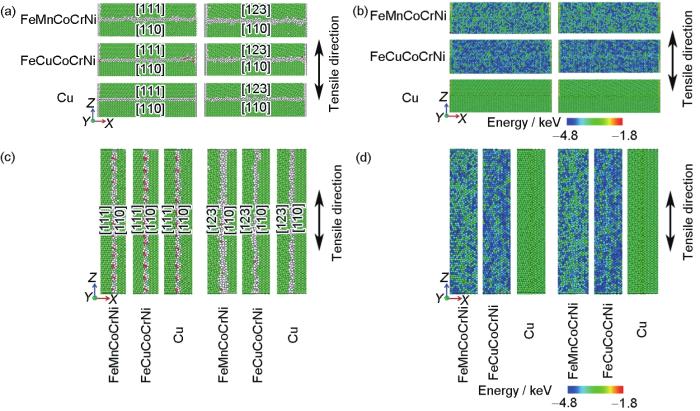
图10
FeMnCoCrNi、FeCuCoCrNi和Cu双晶晶界处的原子结构和原子势能分布
Fig.10
Atomic structures (a, c) and distributions of atomic potential energy (b, d) at grain boundaries for FeMnCoCrNi, FeMnCoCrNi, and pure Cu bicrystals when the grain boundary is perpendicular (a, b) and parallel (c, d) to the tensile direction
当晶界与拉伸方向垂直时,FeMnCoCrNi、FeCuCoCrNi和Cu 3种金属的[111] + [110]和[123] + [110]双晶模型的应力-应变曲线如图11a1~a3所示。工程应变为5%和20%时[111] + [110]和[123] + [110]双晶模型的原子结构分别如图11b1~b3和c1~c3所示。在同一取向组合下,Cu在弹塑性转变点处的应力最大,其次为FeCuCoCrNi和FeMnCoCrNi合金。这归因于高熵合金中较大的晶格畸变,且与FeCuCoCrNi (非稳定层错能为278 mJ/m2)和Cu (非稳定层错能为305 mJ/m2)相比,FeMnCoCrNi的非稳定层错能(222 mJ/m2)最小所致。当取向组合为[111] + [110]时,由于晶格畸变导致位错易于在高熵合金的晶界处形核且大量位错向[111]晶粒内滑移(图4d1),因此,与[123] + [110]取向组合相比,[111] + [110]取向组合FeMnCoCrNi和FeCuCoCrNi合金在弹塑性转变点处的应力较小。对于纯Cu而言,由于无晶格畸变,位错仅在晶界处被激发并向晶界两侧滑移,因此2种取向组合的模型中位错开动的临界应力相近。
图11

图11
晶界与拉伸方向垂直时FeMnCoCrNi、FeCuCoCrNi和Cu双晶的应力-应变曲线及原子结构
Fig.11
Stress-strain curves (a1-a3), atomic structures of the [111] + [110] (b1-b3) and [123] + [110] (c1-c3) bicrystals when ε = 5% and ε = 20% of FeMnCoCrNi (a1-c1), FeCuCoCrNi (a2-c2), and Cu (a3-c3) bicrystals when the grain boundary is perpendicular to the tensile direction
在塑性变形阶段,选取2个特征载荷点进行变形机制的细致分析。当工程应变为5%时,对于[111] + [110]取向组合的双晶而言,3种材料的应力均呈现显著下降的趋势。在FeMnCoCrNi (层错能为-45 mJ/m2)和FeCuCoCrNi (层错能为62 mJ/m2)的[111]晶粒中形成了大量ε-马氏体和层错,而在[110]晶粒中则主要为层错。在纯Cu (层错能为41 mJ/m2)双晶中则仅形成了层错。当双晶模型的取向组合为[123] + [110]时,3种金属的应力同样出现了不同程度地下降。在FeMnCoCrNi的[123]和[110]晶粒中均开动了层错,而在FeCuCoCrNi和Cu中仅在[110]晶粒中识别到层错。当外加应变增加至20%时,相对于其他模型而言,FeMnCoCrNi和FeCuCoCrNi的[111] + [110]模型呈现显著的加工硬化,这对应于这2个模型中较多的ε-马氏体、层错以及少量孪晶的形成。当模型取向组合为[123] + [110]时,在FeCuCoCrNi和Cu双晶的晶界处均发生了断裂,这是由Cu原子较高的势能引起的纯Cu及含Cu合金晶界的能量升高(图10b和d)且[123] + [110]双晶模型中晶界处的原子排列粗糙(图10a)所致。
当晶界与拉伸方向平行时,与图11的结果相似,3种材料不同双晶模型的应力-应变曲线以及特征载荷处的原子结构如图12所示。在同一取向组合下,虽然FeCuCoCrNi和Cu的晶格畸变不同,但2者的非稳定层错能相近。在弹塑性转变点处FeCuCoCrNi和Cu的应力相近且高于FeMnCoCrNi。这表明,对不同金属而言,当晶界平行于变形方向时,位错开动所需的临界应力主要取决于非稳定层错能。而对于同种金属,不同取向组合的模型中位错开动的临界应力相近。在塑性变形阶段,与[123] + [110]相比,3种金属[111] + [110]双晶模型的流变应力较高,同时伴随有较明显的应力波动。而当取向为[111] + [110]时,与FeCuCoCrNi和Cu相比,在50%应变后FeMnCoCrNi的应力下降显著。
图12

图12
晶界与拉伸方向平行时FeMnCoCrNi、FeCuCoCrNi和Cu双晶的应力-应变曲线及原子结构
Fig.12
Stress-strain curves (a1-a3), atomic structures of the [111] + [110] (b1-b3) and [123] + [110] (c1-c3) bicrystals when ε = 5%, ε = 20%, and ε = 100% of FeMnCoCrNi (a1-c1), FeCuCoCrNi (a2-c2), and Cu (a3-c3) bicrystals when the grain boundary is parallel to the tensile direction
进入塑性变形后,当工程应变为5%时,在3种金属不同取向组合的双晶模型中位错滑移后形成的层错为主要变形系统。当继续加载至20%应变时,在[111] + [110]取向组合的FeMnCoCrNi双晶中,以及此取向下FeCuCoCrNi和Cu的[110]晶粒中发生了显著的ε-马氏体相变,这导致宏观应力的增大。而在[123] + [110]模型中,3种金属的主要变形系统均为层错,因此未导致应力随变形增加显著提升。当应变为100%时,在取向组合为[111] + [110]的FeMnCoCrNi中发生了颈缩,晶界严重弯曲,部分原子出现了非晶化,同时还伴随有部分区域发生了逆相变。颈缩导致FeMnCoCrNi在变形后期的应力下降。而在FeCuCoCrNi的[110]晶粒和Cu的[111]、[110]晶粒中,逆相变行为更加显著且变形更加均匀,因此双晶模型整体的流变应力较高。当模型取向组合为[123] + [110]时,由于FeMnCoCrNi的层错能较低,因此合金中开动了多种变形系统。在FeCuCoCrNi的[123]和[110]晶粒中以及Cu的[110]晶粒中发生了层错的消退。3种金属均发生非均匀塑性变形,因此变形结束后的应力呈现略微下降的趋势。通过将FeMnCoCrNi、FeCuCoCrNi和纯Cu 3种材料双晶模型的变形进行对比,发现在相同取向关系和拉伸方向下,较传统低熵材料而言,高熵合金中较大的晶格畸变使晶界更加粗糙,这使得在一定外加载荷作用下,位错易于在高熵合金中形核,且层错能亦对高熵合金的位错行为和马氏体相变有重要影响。
综上所述,本工作系统研究了纳米尺度下不同初始取向组合的高熵双晶在单轴拉伸载荷下力学性能及微观变形机制的异同,同时还通过与低熵金属材料对比,揭示了金属的本征属性(如层错能和晶格畸变)对高熵合金双晶微观力学行为的影响,该工作丰富了高熵合金的塑性变形理论,并可为微纳米尺度多晶金属材料的微结构和取向设计提供理论指导。
3 结论
(1) 对FeMnCoCrNi高熵合金双晶模型而言,在弹塑性转变点处,位错优先在晶界处形核并向两侧的晶粒内滑移。在塑性变形过程中,晶界发生了不同程度的宽化和弯曲,双晶模型中各晶粒的变形系统开动与相同取向单晶的模拟结果一致。当晶界与变形方向垂直时,颈缩易于在晶界处发生,这导致模型整体的流变应力下降。而当晶界与变形方向平行时,模型在整个塑性变形过程中保持1 GPa以上的流变应力。
(2) 在FeMnCoCrNi高熵合金的双晶模型中,[111]与[110]取向组合的双晶流变应力波动幅度最大,同时呈现出最强的加工硬化能力。其中,应力的下降归因于大量的位错发生了滑移,而硬化则是由较多的ε-马氏体、层错以及孪晶形成所致。
(3) 当晶界与变形方向垂直时,与Cu相比,FeMnCoCrNi和FeCuCoCrNi高熵合金中的晶格畸变使晶界更加粗糙,这使得在相同载荷条件下高熵合金中的位错易于形核,且层错能较低的FeMnCoCrNi中形成的ε-马氏体最多。由于Cu原子的势能较高,晶界的稳定性降低,从而导致FeCuCoCrNi和Cu在晶界位置发生了断裂。当晶界与变形方向平行时,3种金属弹塑性转变点处的应力主要取决于非稳定层错能的大小。
参考文献
Nanostructured high-entropy alloys with multiple principal elements: Novel alloy design concepts and outcomes
[J].
Mechanical properties, microstructure and thermal stability of a nanocrystalline CoCrFeMnNi high-entropy alloy after severe plastic deformation
[J].
Effect of Ti content on microstructure and properties of Ti x ZrVNb refractory high-entropy alloys
[J].
A fracture-resistant high-entropy alloy for cryogenic applications
[J].
High-entropy alloys are equiatomic, multi-element systems that can crystallize as a single phase, despite containing multiple elements with different crystal structures. A rationale for this is that the configurational entropy contribution to the total free energy in alloys with five or more major elements may stabilize the solid-solution state relative to multiphase microstructures. We examined a five-element high-entropy alloy, CrMnFeCoNi, which forms a single-phase face-centered cubic solid solution, and found it to have exceptional damage tolerance with tensile strengths above 1 GPa and fracture toughness values exceeding 200 MPa·m(1/2). Furthermore, its mechanical properties actually improve at cryogenic temperatures; we attribute this to a transition from planar-slip dislocation activity at room temperature to deformation by mechanical nanotwinning with decreasing temperature, which results in continuous steady strain hardening. Copyright © 2014, American Association for the Advancement of Science.
Microstructure and wear behavior of Al x Co1.5CrFeNi1.5Ti y high-entropy alloys
[J].
Corrosion resistant nanostructured eutectic high entropy alloy
[J].
Metastable high-entropy dual-phase alloys overcome the strength-ductility trade-off
[J].
Multi-heterostructure and mechanical properties of N-doped FeMnCoCr high entropy alloy
[J].
Enhanced strength and ductility in a high-entropy alloy via ordered oxygen complexes
[J].
Alloy design strategies and future trends in high-entropy alloys
[J].
Lattice distortion in high-entropy alloys
[J].Lattice distortion has been deemed as one of the most distinctive structural features of high-entropy alloys. However, despite its fundamental importance, the notion of lattice distortion and its characterization is still an issue under debate. In this work, based on the recent work on theoretical modeling, atomistic simulations and experiments, the physical origin of lattice distortion in high-entropy alloys and its resultant intrinsic residual stresses or strains are discussed.
高熵合金中的晶格畸变
[J].
A critical review of high entropy alloys and related concepts
[J].
Short-range order and its impact on the CrCoNi medium-entropy alloy
[J].
Strengthening in multi-principal element alloys with local-chemical-order roughened dislocation pathways
[J].High-entropy and medium-entropy alloys are presumed to have a configurational entropy as high as that of an ideally mixed solid solution (SS) of multiple elements in near-equal proportions. However, enthalpic interactions inevitably render such chemically disordered SSs rare and metastable, except at very high temperatures. Here we highlight the wide variety of local chemical ordering (LCO) that sets these concentrated SSs apart from traditional solvent-solute ones. Using atomistic simulations, we reveal that the LCO of the multi-principal-element NiCoCr SS changes with alloy processing conditions, producing a wide range of generalized planar fault energies. We show that the LCO heightens the ruggedness of the energy landscape and raises activation barriers governing dislocation activities. This influences the selection of dislocation pathways in slip, faulting, and twinning, and increases the lattice friction to dislocation motion via a nanoscale segment detrapping mechanism. In contrast, severe plastic deformation reduces the LCO towards random SS.
Ultrastrong medium-entropy single-phase alloys designed via severe lattice distortion
[J].
Microstructure and mechanical properties of Cr-rich Co-Cr-Fe-Ni high entropy alloys designed by valence electron concentration
[J].
Synergy effect of multi-strengthening mechanisms in FeMnCoCrN HEA at cryogenic temperature
[J].
Microstructure and mechanical properties of a FeMnCoCr high-entropy alloy with heterogeneous structure
[J].Growing attention has been placed on high-entropy alloys (HEAs) owing to their promising mechanical properties. Particularly, HEAs in which the main crystal structure is fcc are attracting significant attention. Although such alloys exhibit a good combination of strength and ductility, they cannot meet the increasing demands of applications because of limited yield strengths. In recent years, researchers have tried to improve yield strengths of HEAs by refining grains and introducing interstitial atoms. However, the processing cost is high and is often accompanied by the significant loss of ductility. In this study, we propose a simple processing route incorporating cold rolling at medium thickness reductions and short-time annealing at medium temperatures to obtain a heterogeneous structure in Fe-Mn based HEAs consisting of deformed grains with an average diameter of several tens of microns and recrystallized ultrafine grains. By simultaneously introducing multiple strengthening mechanisms, including the strengthening contributed by the microstructural characteristics of dense dislocations, grain refinement, precipitates, ε-martensite, α-martensite, and recovery twins, as well as the strengthening induced by deformation twinning and ε-martensite phase transition that occurs continuously during deformation, the yield strength of the alloy significantly increases compared with that of the fully recrystallized material and reaches 825 MPa. Simultaneously, due to the activation of significant deformation twinning and deformation-induced martensitic transformation, the uniform elongation of the alloy is about 28.6%. The proposed material fabrication method is simple, cost-effective, and can effectively improve the mechanical properties of Fe-Mn based HEAs, providing new insight into optimizing the mechanical properties of low stacking fault energy alloys of the fcc structure.
非均匀组织FeMnCoCr高熵合金的微观结构和力学性能
[J].提出了一种简单的高熵合金加工工艺,即对Fe-Mn系高熵合金采用中等形变量冷轧和中温短时退火相结合的方法,获得了由晶粒尺寸为数十微米的形变晶粒和超细尺度再结晶晶粒组成的非均匀结构。通过向合金中同时引入由高密度位错、晶粒细化、析出相、ε-马氏体、α-马氏体和回复孪晶等微观结构特征及变形过程中持续发生的形变孪生、ε-马氏体相变引起的多种强化机制,使屈服强度较充分再结晶态显著提升并达到825 MPa。同时,在塑性变形过程中由于发生了显著的形变孪生和一定的由形变诱发的奥氏体向ε-马氏体转变,合金仍具有约28.6%的均匀延伸率,合金的综合力学性能得到有效提升。该工艺为优化以fcc结构为主的低层错能合金的力学性能提供了新思路。
Joint contribution of transformation and twinning to the high strength-ductility combination of a FeMnCoCr high entropy alloy at cryogenic temperatures
[J].
Segregation-driven grain boundary spinodal decomposition as a pathway for phase nucleation in a high-entropy alloy
[J].
The effect of Co and Cr substitutions for Ni on mechanical properties and plastic deformation mechanism of FeMnCoCrNi high entropy alloys
[J].The elastic constants, ideal tensile strength (ITS), stacking fault energy (SFE), lattice constant and magnetic moment of FeMnCoCrNi high entropy alloys with varying Co and Cr contents at 0 and 300 K were systematically investigated by first-principle calculations. For the alloys with Co substitution for Ni, at both temperatures the elastic stability of the face-centered cubic (fcc) phase, bulk elastic modulus (B), Young's modulus (E), shear modulus (G) and ITS increase monotonically with increasing Co content. However, the Cauchy pressure (CP), Pugh ratio (B/G), Poisson ratio (v), Zener anisotropy ratio (AZ) and elastic anisotropy ratio (AVR) decrease monotonically. The SFE also decreases with the increase of Co, resulting in the change of plastic deformation mechanism from dislocation slip to mechanical twinning, and then to hcp-martensitic transformation. This elucidates the underlying mechanism of the effect of Co addition on the strength and micromechanical behavior of FeMnCoCrNi alloys. Compared with Co, the Cr substitution for Ni leads to the more complex change of elastic constants and ITS. The increase of Cr shows the similar effect on SFE and deformation mechanism as that of Co. The variation of valence electron concentration and magnetism affect the SFE. The increase of either Co or Cr leads to the reduced magnetic moments of Fe and Mn. This could be responsible for the monotonic decrease of both lattice constant and SFE as the Co content increases. However, for the Cr addition case, multiple factors may affect the evolution of lattice constant and SFE. These findings shed light on the deformation mechanism of the alloys with different compositions.
Reverse transformation in [110]-oriented face-centered-cubic single crystals studied by atomic simulations
[J].
Model interatomic potentials and lattice strain in a high-entropy alloy
[J].
Interstitial atoms enable joint twinning and transformation induced plasticity in strong and ductile high-entropy alloys
[J].High-entropy alloys (HEAs) consisting of multiple principle elements provide an avenue for realizing exceptional mechanical, physical and chemical properties. We report a novel strategy for designing a new class of HEAs incorporating the additional interstitial element carbon. This results in joint activation of twinning-and transformation-induced plasticity (TWIP and TRIP) by tuning the matrix phase's instability in a metastable TRIP-assisted dual-phase HEA. Besides TWIP and TRIP, such alloys benefit from massive substitutional and interstitial solid solution strengthening as well as from the composite effect associated with its dual-phase structure. Nanosize particle formation and grain size reduction are also utilized. The new interstitial TWIP-TRIP-HEA thus unifies all metallic strengthening mechanisms in one material, leading to twice the tensile strength compared to a single-phase HEA with similar composition, yet, at identical ductility.
Orientation-dependent mechanical responses and plastic deformation mechanisms of FeMnCoCrNi high-entropy alloy: A molecular dynamics study
[J].
Stabilizing nanostructures in metals using grain and twin boundary architectures
[J].
Structural metals at extremes
[J].
Mechanical behavior of nanocrystalline metals and alloys
[J].
Atomic simulations of dislocation emission from Cu/Cu and Co/Cu grain boundaries
[J].
Atomic simulation of grain boundary sliding in Co/Cu two-phase bicrystals
[J].
Entropy-induced transition on grain-boundary migration in multi-principal element alloys
[J].
Tensile deformation behavior of twist grain boundaries in CoCrFeMnNi high entropy alloy bicrystals
[J].High entropy alloys (HEA) are a class of materials that consist of multiple elemental species in similar concentrations. The use of elements in far from dilute concentrations introduces a multi-dimensional composition design space by which the properties of metallic systems can be tailored. While the mechanical behavior of HEAs has been the subject of active research recently, the role of grain boundaries (GBs) in their deformation behavior remains poorly understood. Motivated by recent experiments on HEAs demonstrating that GBs act as nucleation sites for deformation twins, herein, we leverage atomistic simulations to construct a series of equiatomic CoCrFeMnNi HEA bicrystals with [Formula: see text] and [Formula: see text] symmetric twist GBs and examine their tensile behavior and underlying deformation mechanisms at 77 K. Simulation results reveal that plastic deformation proceeds by the nucleation of partial dislocations from GBs, which then grow with further loading by bowing into the bulk crystals leaving behind stacking faults. Variations in the nucleation stress exist as function of GB character, defined in this work by the twist angle. Our results provide future avenues to explore GBs as a microstructure design tool to develop HEAs with tailored properties.
Understanding the physical metallurgy of the CoCrFeMnNi high-entropy alloy: An atomistic simulation study
[J].Although high-entropy alloys (HEAs) are attracting interest, the physical metallurgical mechanisms related to their properties have mostly not been clarified, and this limits wider industrial applications, in addition to the high alloy costs. We clarify the physical metallurgical reasons for the materials phenomena (sluggish diffusion and micro-twining at cryogenic temperatures) and investigate the effect of individual elements on solid solution hardening for the equiatomic CoCrFeMnNi HEA based on atomistic simulations (Monte Carlo, molecular dynamics and molecular statics). A significant number of stable vacant lattice sites with high migration energy barriers exists and is thought to cause the sluggish diffusion. We predict that the hexagonal close-packed (hcp) structure is more stable than the face-centered cubic (fcc) structure at 0 K, which we propose as the fundamental reason for the micro-twinning at cryogenic temperatures. The alloying effect on the critical resolved shear stress (CRSS) is well predicted by the atomistic simulation, used for a design of non-equiatomic fcc HEAs with improved strength, and is experimentally verified. This study demonstrates the applicability of the proposed atomistic approach combined with a thermodynamic calculation technique to a computational design of advanced HEAs.
A semi-empirical interatomic potential for the Cu-Ti binary system
[J].
Generalized stacking fault energies of alloys
[J].
Fast parallel algorithms for short-range molecular dynamics
[J].
Sualization and analysis of atomistic simulation data with OVITO—The open visualization tool
[J].
Structural characterization of deformed crystals by analysis of common atomic neighborhood
[J].
Extracting dislocations and non-dislocation crystal defects from atomistic simulation data
[J].
Temperature dependent stacking fault energy of FeCrCoNiMn high entropy alloy
[J].
An overview of modeling the stacking faults in lightweight and high-entropy alloys: Theory and application
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}