1
2012
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
1
2012
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
几种高温材料抗燃气烧蚀性能的研究
1
1995
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
几种高温材料抗燃气烧蚀性能的研究
1
1995
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
星际航行等离子火箭发动机
1
1964
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
星际航行等离子火箭发动机
1
1964
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
Ductility and impact resistance of powder-metallurgical molybdenum-rhenium alloys
1
2006
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
Properties of tungsten-rhenium and tungsten-rhenium with hafnium carbide
1
2009
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
The alloys of rhenium with molybdenum or with tungsten and having good high-temperature properties
1
1956
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
Ultrahigh temperature tensile properties of arc-melted tungsten and tungsten-iridium alloys
2
1991
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
... W-Ir二元合金和纯W[35~41]的弹性常数(C11、C12、C44)、弹性模量(B、G、E)、G / B和Cp随Ir添加量的变化曲线示于图4.可以看到,纯W计算值均在其他实验和计算所得结果范围之内,表明了当前弹性系数计算方法和参数设置的可靠性.弹性系数C11表征了晶体材料在承受单轴应变时的抗变形能力,C12和C44则分别表征了晶体材料沿着(00l) (l为晶面指数)侧面和<111>四度轴向的抗剪切能力.图4表明,随着Ir含量的增加,C11和C44呈减小趋势,表明Ir的加入可以明显降低单轴受力和<111>轴向剪切时的抗变形能力,提升材料韧性;而C12则随Ir含量增加而单调增加,表明合金抗平面剪切变形能力增加,预示该方向变形时材料刚度增加;弹性模量B、G和E均随着Ir的添加单调递减,表明Ir的添加会降低材料发生体积变形、剪切变形和单轴变形时的抗变形能力.这一结果预示W-Ir合金在遭受外部应力时,W-Ir合金体系比纯W体系更易达到剪切应力极限诱发位错发射,通过塑性协变缓解裂纹尖端应力集中,稳定了裂纹尖端(韧性特征),避免或者延缓了脆性断裂.这种Ir诱发的易变形特性也从另一个角度验证了图2中形成能的计算结果,即Ir能诱发相的不稳定特性.随着Ir含量从0增加到12.96%,表征韧脆特征的参量G / B从0.52 (脆性)降低到0.37 (韧性);Cp为正值,且随着Ir含量的增加从42 GPa增加到112 GPa,也预示着Ir的添加能改善W基体的脆性,提升其韧性[18].这一结论与Luo等[8,9]和McKamey等[10]的实验观察一致.该实验研究发现,Ir的加入会导致钨合金的极限拉伸应力和屈服强度下降,韧性增加.另外,对比Ir和Mo在W中的韧化效果,Mo含量从0变化至70%,G / B仅降低0.03[49],而Ir含量从0变化到12.96%,G / B降低0.15,可见Ir对W的韧化效果远远大于Mo对W的韧化作用.本工作将进一步从电子价键的角度去分析其韧化机理. ...
Solution softening mechanism of iridium and rhenium in tungsten at room temperature
2
1991
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
... W-Ir二元合金和纯W[35~41]的弹性常数(C11、C12、C44)、弹性模量(B、G、E)、G / B和Cp随Ir添加量的变化曲线示于图4.可以看到,纯W计算值均在其他实验和计算所得结果范围之内,表明了当前弹性系数计算方法和参数设置的可靠性.弹性系数C11表征了晶体材料在承受单轴应变时的抗变形能力,C12和C44则分别表征了晶体材料沿着(00l) (l为晶面指数)侧面和<111>四度轴向的抗剪切能力.图4表明,随着Ir含量的增加,C11和C44呈减小趋势,表明Ir的加入可以明显降低单轴受力和<111>轴向剪切时的抗变形能力,提升材料韧性;而C12则随Ir含量增加而单调增加,表明合金抗平面剪切变形能力增加,预示该方向变形时材料刚度增加;弹性模量B、G和E均随着Ir的添加单调递减,表明Ir的添加会降低材料发生体积变形、剪切变形和单轴变形时的抗变形能力.这一结果预示W-Ir合金在遭受外部应力时,W-Ir合金体系比纯W体系更易达到剪切应力极限诱发位错发射,通过塑性协变缓解裂纹尖端应力集中,稳定了裂纹尖端(韧性特征),避免或者延缓了脆性断裂.这种Ir诱发的易变形特性也从另一个角度验证了图2中形成能的计算结果,即Ir能诱发相的不稳定特性.随着Ir含量从0增加到12.96%,表征韧脆特征的参量G / B从0.52 (脆性)降低到0.37 (韧性);Cp为正值,且随着Ir含量的增加从42 GPa增加到112 GPa,也预示着Ir的添加能改善W基体的脆性,提升其韧性[18].这一结论与Luo等[8,9]和McKamey等[10]的实验观察一致.该实验研究发现,Ir的加入会导致钨合金的极限拉伸应力和屈服强度下降,韧性增加.另外,对比Ir和Mo在W中的韧化效果,Mo含量从0变化至70%,G / B仅降低0.03[49],而Ir含量从0变化到12.96%,G / B降低0.15,可见Ir对W的韧化效果远远大于Mo对W的韧化作用.本工作将进一步从电子价键的角度去分析其韧化机理. ...
Grain growth behaviour and high strain rate tensile properties of gas tungsten arc welds in iridium alloy DOP-26
2
2000
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
... W-Ir二元合金和纯W[35~41]的弹性常数(C11、C12、C44)、弹性模量(B、G、E)、G / B和Cp随Ir添加量的变化曲线示于图4.可以看到,纯W计算值均在其他实验和计算所得结果范围之内,表明了当前弹性系数计算方法和参数设置的可靠性.弹性系数C11表征了晶体材料在承受单轴应变时的抗变形能力,C12和C44则分别表征了晶体材料沿着(00l) (l为晶面指数)侧面和<111>四度轴向的抗剪切能力.图4表明,随着Ir含量的增加,C11和C44呈减小趋势,表明Ir的加入可以明显降低单轴受力和<111>轴向剪切时的抗变形能力,提升材料韧性;而C12则随Ir含量增加而单调增加,表明合金抗平面剪切变形能力增加,预示该方向变形时材料刚度增加;弹性模量B、G和E均随着Ir的添加单调递减,表明Ir的添加会降低材料发生体积变形、剪切变形和单轴变形时的抗变形能力.这一结果预示W-Ir合金在遭受外部应力时,W-Ir合金体系比纯W体系更易达到剪切应力极限诱发位错发射,通过塑性协变缓解裂纹尖端应力集中,稳定了裂纹尖端(韧性特征),避免或者延缓了脆性断裂.这种Ir诱发的易变形特性也从另一个角度验证了图2中形成能的计算结果,即Ir能诱发相的不稳定特性.随着Ir含量从0增加到12.96%,表征韧脆特征的参量G / B从0.52 (脆性)降低到0.37 (韧性);Cp为正值,且随着Ir含量的增加从42 GPa增加到112 GPa,也预示着Ir的添加能改善W基体的脆性,提升其韧性[18].这一结论与Luo等[8,9]和McKamey等[10]的实验观察一致.该实验研究发现,Ir的加入会导致钨合金的极限拉伸应力和屈服强度下降,韧性增加.另外,对比Ir和Mo在W中的韧化效果,Mo含量从0变化至70%,G / B仅降低0.03[49],而Ir含量从0变化到12.96%,G / B降低0.15,可见Ir对W的韧化效果远远大于Mo对W的韧化作用.本工作将进一步从电子价键的角度去分析其韧化机理. ...
铱及铱铑合金的高温氧化性能研究
1
2018
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
铱及铱铑合金的高温氧化性能研究
1
2018
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
塑性铱的研究及其应用
1
1999
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
塑性铱的研究及其应用
1
1999
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
Peculiarities of defect structure and mechanical properties of iridium: Results of ab initio electronic structure calculations
1
2000
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
Modeling the dislocation properties and mechanical behavior of Ir, Rh, and their refractory alloys
1
2005
... 难熔金属W因为具有高熔点(3410℃)、高硬度、高强度、高密度(19.32 g/cm3)、良好的耐化学腐蚀性和抗烧蚀性等特点,可用作能源、冶金、化工、电子等民用领域,以及国防军工、航空航天、新能源、核工业等尖端科技领域的关键支撑材料[1].其中,在航空航天领域应用的固体火箭燃料发动机非冷却式喷管中所用的W,需具有耐高温、抗热震和耐侵蚀等特性,使其在高温条件下服役较长时间而不失效[2~4].但纯W具有低温脆性易裂、不易加工以及高温强度不足的问题,抗蚀性也有待加强.固溶合金化方法可以有效改善这些问题[5,6].如Re元素自从1956年被Geach和Hughes[7]发现能改善W的塑性以来,在各种成分范围(0~50%,原子分数)内的合金化作用均已被广泛研究.与Re具有相似电子结构的Ir也引起研究者们的关注.贵金属Ir密度略低于Os,熔点高(2447℃)、硬度高、弹性模量高,具有极强的耐腐蚀性、热稳定性及高温强度等性能.就化学稳定性而言,它是金属中最耐腐蚀的元素,不溶于所有的无机酸,包括王水,能经受许多熔融试剂和高温硅酸盐的侵蚀.Ir的这些物化性能使它能在非常苛刻的条件下工作,在传统高温材料领域获得应用,适合添加到钨基合金中使其能更好地在高温特殊环境下服役.Luo等[8,9]分别研究了W-0.4%Ir和W-0.8%Ir (质量分数)中Ir含量对W的室温以及高温力学性能的影响,并与Re做对比.研究表明,Ir对W的室温韧化性能可以与Re相媲美,而且可以较好地维持W的高温强度.McKamey等[10]发现,0.3%Ir (质量分数)可以提升钨合金韧性的程度为9%~15%.但让研究者费解的是,Ir具有fcc结构,然而却具有不同于常规fcc结构的力学性能,比较脆,一般认为只有在1600℃以上才具有良好的塑性[11,12].研究表明,Ir具有更多的价电子因此导致其价键具有明显的方向性[13],其金属键具有伪共价(pseudo-covalent)特征[14].具有脆性特征的Ir元素能较好地起到韧化钨合金的作用,这种奇妙特性转变的内在影响因素和作用机制尚未揭示.除此之外,目前关于Ir的添加量和其固溶合金化所带来的力学以及热力学性能方面的变化关系尚未建立.因此从微观尺度上研究Ir的加入对钨基合金性能的影响非常必要,也具有重要意义,有助于研究者们更好地基于Ir对W的影响特性设计W-Ir合金的成分,拓展钨基合金在高温领域的应用. ...
Crystal structure and stability of complex precipitate phases in Al-Cu-Mg-(Si) and Al-Zn-Mg alloys
1
2001
... 形成能(ΔE)主要用于表征固溶合金的相对稳定性,是衡量合金元素对基体材料影响的重要参数.计算公式如下[15]: ...
Calculations of single-crystal elastic constants made simple
1
2010
... 弹性系数可以反映材料的力学性能.本工作采用通用线性独立耦合应变(universal linear-independent coupling strains,ULICS)方法[16]计算合金的弹性常数.该方法中,应变在应变空间中线性无关,所有的应力分量和弹性常数耦合.在拟合变形晶体的应变-应力关系的基础上,利用奇异值分解进行线性最小二乘拟合,得到弹性常数矩阵( Cij )[17].由于在工程应用中,考虑更多的是弹性模量,如Young's模量(E)、体积弹性模量(B)、剪切模量(G)等[18],因此通过Voigt-Reuss方案[19]计算B、G、E等参量: ...
1
2007
... 弹性系数可以反映材料的力学性能.本工作采用通用线性独立耦合应变(universal linear-independent coupling strains,ULICS)方法[16]计算合金的弹性常数.该方法中,应变在应变空间中线性无关,所有的应力分量和弹性常数耦合.在拟合变形晶体的应变-应力关系的基础上,利用奇异值分解进行线性最小二乘拟合,得到弹性常数矩阵( Cij )[17].由于在工程应用中,考虑更多的是弹性模量,如Young's模量(E)、体积弹性模量(B)、剪切模量(G)等[18],因此通过Voigt-Reuss方案[19]计算B、G、E等参量: ...
Ab initio calculations of mechanical properties: Methods and applications
2
2015
... 弹性系数可以反映材料的力学性能.本工作采用通用线性独立耦合应变(universal linear-independent coupling strains,ULICS)方法[16]计算合金的弹性常数.该方法中,应变在应变空间中线性无关,所有的应力分量和弹性常数耦合.在拟合变形晶体的应变-应力关系的基础上,利用奇异值分解进行线性最小二乘拟合,得到弹性常数矩阵( Cij )[17].由于在工程应用中,考虑更多的是弹性模量,如Young's模量(E)、体积弹性模量(B)、剪切模量(G)等[18],因此通过Voigt-Reuss方案[19]计算B、G、E等参量: ...
... W-Ir二元合金和纯W[35~41]的弹性常数(C11、C12、C44)、弹性模量(B、G、E)、G / B和Cp随Ir添加量的变化曲线示于图4.可以看到,纯W计算值均在其他实验和计算所得结果范围之内,表明了当前弹性系数计算方法和参数设置的可靠性.弹性系数C11表征了晶体材料在承受单轴应变时的抗变形能力,C12和C44则分别表征了晶体材料沿着(00l) (l为晶面指数)侧面和<111>四度轴向的抗剪切能力.图4表明,随着Ir含量的增加,C11和C44呈减小趋势,表明Ir的加入可以明显降低单轴受力和<111>轴向剪切时的抗变形能力,提升材料韧性;而C12则随Ir含量增加而单调增加,表明合金抗平面剪切变形能力增加,预示该方向变形时材料刚度增加;弹性模量B、G和E均随着Ir的添加单调递减,表明Ir的添加会降低材料发生体积变形、剪切变形和单轴变形时的抗变形能力.这一结果预示W-Ir合金在遭受外部应力时,W-Ir合金体系比纯W体系更易达到剪切应力极限诱发位错发射,通过塑性协变缓解裂纹尖端应力集中,稳定了裂纹尖端(韧性特征),避免或者延缓了脆性断裂.这种Ir诱发的易变形特性也从另一个角度验证了图2中形成能的计算结果,即Ir能诱发相的不稳定特性.随着Ir含量从0增加到12.96%,表征韧脆特征的参量G / B从0.52 (脆性)降低到0.37 (韧性);Cp为正值,且随着Ir含量的增加从42 GPa增加到112 GPa,也预示着Ir的添加能改善W基体的脆性,提升其韧性[18].这一结论与Luo等[8,9]和McKamey等[10]的实验观察一致.该实验研究发现,Ir的加入会导致钨合金的极限拉伸应力和屈服强度下降,韧性增加.另外,对比Ir和Mo在W中的韧化效果,Mo含量从0变化至70%,G / B仅降低0.03[49],而Ir含量从0变化到12.96%,G / B降低0.15,可见Ir对W的韧化效果远远大于Mo对W的韧化作用.本工作将进一步从电子价键的角度去分析其韧化机理. ...
On the elastic moduli of a crystal and voigt and reuss relations
1
1965
... 弹性系数可以反映材料的力学性能.本工作采用通用线性独立耦合应变(universal linear-independent coupling strains,ULICS)方法[16]计算合金的弹性常数.该方法中,应变在应变空间中线性无关,所有的应力分量和弹性常数耦合.在拟合变形晶体的应变-应力关系的基础上,利用奇异值分解进行线性最小二乘拟合,得到弹性常数矩阵( Cij )[17].由于在工程应用中,考虑更多的是弹性模量,如Young's模量(E)、体积弹性模量(B)、剪切模量(G)等[18],因此通过Voigt-Reuss方案[19]计算B、G、E等参量: ...
The elastic behaviour of a crystalline aggregate
1
1952
... 式中,BV、GV和BR、GR分别为按照Voigt和Reuss方法所得体积模量和剪切模量;Cij 、Sij 分别是弹性系数和应变系数.由Voigt-Reuss-Hill[20]近似得到: ...
XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals
1
1954
... 另外,采用Pugh值(G / B)和Cauchy应力Cp= (C12 - C44)来衡量W-Ir合金的韧脆性变化[21].G / B <0.5预示材料呈现韧性,而相反地,G / B > 0.5预示材料呈现脆性.Cp > 0预示材料呈现韧性,Cp < 0预示材料呈现脆性. ...
First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures
1
2008
... 考虑到钨合金的高温应用背景,有必要研究W-Ir合金的热力学稳定性.本工作中W-Ir合金的热力学性能采用第一原理平面波赝势方法VASP (Vienna Ab-initio Simulation Package)软件包结合晶体结构声子计算软件包Phonopy[22]进行计算.该计算基于密度泛函微扰理论,包含了格点振动过程中的非简谐效应,先计算格点振动的声子谱,进一步计算得到自由能(F)、熵(S)、焓(H)、比热容(cV )等[23]热力学性能参量,以考察W-Ir合金的热稳定性变化: ...
First-principles study of lattice dynamics and thermodynamics of TiO2 polymorphs
1
2011
... 考虑到钨合金的高温应用背景,有必要研究W-Ir合金的热力学稳定性.本工作中W-Ir合金的热力学性能采用第一原理平面波赝势方法VASP (Vienna Ab-initio Simulation Package)软件包结合晶体结构声子计算软件包Phonopy[22]进行计算.该计算基于密度泛函微扰理论,包含了格点振动过程中的非简谐效应,先计算格点振动的声子谱,进一步计算得到自由能(F)、熵(S)、焓(H)、比热容(cV )等[23]热力学性能参量,以考察W-Ir合金的热稳定性变化: ...
Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets
4
2011
... 为了进一步探究相稳定、力学及热力学性质产生的物理根源,有必要研究基体元素和添加元素之间的价键作用.但是基于投影缀加波(PAW)交换关联势的密度泛函理论方法得到的电子结构无法直接给出原子之间的价键作用信息,因此本工作进一步计算了投影晶体轨道Hamilton布局(projected crystal orbital Hamiltonian population,pCOHP)[24],对原子轨道电子重叠情况进行分析.pCOHP是一种将密度泛函理论所得到的能带波函数原子轨道化处理的方法,是Deringer等[24]在1983年Hughbanks和Hoffmann[25]提出的晶体轨道重合布局(crystal orbital overlap population,COOP)和1993年Dronskowski和Blöch[26]发展的晶体轨道Hamilton布局(crystal orbital Hamilton population,COHP)分析的基础上进一步发展起来的.在pCOHP方法中,依次对能带波函数进行转移矩阵(包含单电子轨道函数)处理、投影密度矩阵(包含基于原子轨道线性组合获得的相关原子轨道化学信息)处理、Hamilton矩阵化处理,最后将该Hamilton布局权重作用到态密度曲线上,所得即pCOHP曲线[24].曲线包含成键和反键轨道电子信息,成键轨道电子引起能量下降,而反键轨道电子引起能量增加,可以定量表征键强,适用于共价键、离子键、金属键以及分子键体系[24].本工作中,pCOHP曲线基于面向电子结构重建的局域轨道基组(local orbital suite towards electronic-structure reconstruction,LOBSTER)程序[27]计算获得.除此之外,本工作利用二次差分电荷密度来分析掺杂前后电子的转移以及空间分布情况. ...
... [24]在1983年Hughbanks和Hoffmann[25]提出的晶体轨道重合布局(crystal orbital overlap population,COOP)和1993年Dronskowski和Blöch[26]发展的晶体轨道Hamilton布局(crystal orbital Hamilton population,COHP)分析的基础上进一步发展起来的.在pCOHP方法中,依次对能带波函数进行转移矩阵(包含单电子轨道函数)处理、投影密度矩阵(包含基于原子轨道线性组合获得的相关原子轨道化学信息)处理、Hamilton矩阵化处理,最后将该Hamilton布局权重作用到态密度曲线上,所得即pCOHP曲线[24].曲线包含成键和反键轨道电子信息,成键轨道电子引起能量下降,而反键轨道电子引起能量增加,可以定量表征键强,适用于共价键、离子键、金属键以及分子键体系[24].本工作中,pCOHP曲线基于面向电子结构重建的局域轨道基组(local orbital suite towards electronic-structure reconstruction,LOBSTER)程序[27]计算获得.除此之外,本工作利用二次差分电荷密度来分析掺杂前后电子的转移以及空间分布情况. ...
... [24].曲线包含成键和反键轨道电子信息,成键轨道电子引起能量下降,而反键轨道电子引起能量增加,可以定量表征键强,适用于共价键、离子键、金属键以及分子键体系[24].本工作中,pCOHP曲线基于面向电子结构重建的局域轨道基组(local orbital suite towards electronic-structure reconstruction,LOBSTER)程序[27]计算获得.除此之外,本工作利用二次差分电荷密度来分析掺杂前后电子的转移以及空间分布情况. ...
... [24].本工作中,pCOHP曲线基于面向电子结构重建的局域轨道基组(local orbital suite towards electronic-structure reconstruction,LOBSTER)程序[27]计算获得.除此之外,本工作利用二次差分电荷密度来分析掺杂前后电子的转移以及空间分布情况. ...
Chains of trans-edge-sharing molybdenum octahedra: Metal-metal bonding in extended systems
1
1983
... 为了进一步探究相稳定、力学及热力学性质产生的物理根源,有必要研究基体元素和添加元素之间的价键作用.但是基于投影缀加波(PAW)交换关联势的密度泛函理论方法得到的电子结构无法直接给出原子之间的价键作用信息,因此本工作进一步计算了投影晶体轨道Hamilton布局(projected crystal orbital Hamiltonian population,pCOHP)[24],对原子轨道电子重叠情况进行分析.pCOHP是一种将密度泛函理论所得到的能带波函数原子轨道化处理的方法,是Deringer等[24]在1983年Hughbanks和Hoffmann[25]提出的晶体轨道重合布局(crystal orbital overlap population,COOP)和1993年Dronskowski和Blöch[26]发展的晶体轨道Hamilton布局(crystal orbital Hamilton population,COHP)分析的基础上进一步发展起来的.在pCOHP方法中,依次对能带波函数进行转移矩阵(包含单电子轨道函数)处理、投影密度矩阵(包含基于原子轨道线性组合获得的相关原子轨道化学信息)处理、Hamilton矩阵化处理,最后将该Hamilton布局权重作用到态密度曲线上,所得即pCOHP曲线[24].曲线包含成键和反键轨道电子信息,成键轨道电子引起能量下降,而反键轨道电子引起能量增加,可以定量表征键强,适用于共价键、离子键、金属键以及分子键体系[24].本工作中,pCOHP曲线基于面向电子结构重建的局域轨道基组(local orbital suite towards electronic-structure reconstruction,LOBSTER)程序[27]计算获得.除此之外,本工作利用二次差分电荷密度来分析掺杂前后电子的转移以及空间分布情况. ...
Crystal orbital Hamilton populations (COHP): Energy-resolved visualization of chemical bonding in solids based on density-functional calculations
1
1993
... 为了进一步探究相稳定、力学及热力学性质产生的物理根源,有必要研究基体元素和添加元素之间的价键作用.但是基于投影缀加波(PAW)交换关联势的密度泛函理论方法得到的电子结构无法直接给出原子之间的价键作用信息,因此本工作进一步计算了投影晶体轨道Hamilton布局(projected crystal orbital Hamiltonian population,pCOHP)[24],对原子轨道电子重叠情况进行分析.pCOHP是一种将密度泛函理论所得到的能带波函数原子轨道化处理的方法,是Deringer等[24]在1983年Hughbanks和Hoffmann[25]提出的晶体轨道重合布局(crystal orbital overlap population,COOP)和1993年Dronskowski和Blöch[26]发展的晶体轨道Hamilton布局(crystal orbital Hamilton population,COHP)分析的基础上进一步发展起来的.在pCOHP方法中,依次对能带波函数进行转移矩阵(包含单电子轨道函数)处理、投影密度矩阵(包含基于原子轨道线性组合获得的相关原子轨道化学信息)处理、Hamilton矩阵化处理,最后将该Hamilton布局权重作用到态密度曲线上,所得即pCOHP曲线[24].曲线包含成键和反键轨道电子信息,成键轨道电子引起能量下降,而反键轨道电子引起能量增加,可以定量表征键强,适用于共价键、离子键、金属键以及分子键体系[24].本工作中,pCOHP曲线基于面向电子结构重建的局域轨道基组(local orbital suite towards electronic-structure reconstruction,LOBSTER)程序[27]计算获得.除此之外,本工作利用二次差分电荷密度来分析掺杂前后电子的转移以及空间分布情况. ...
LOBSTER: A tool to extract chemical bonding from plane-wave based DFT
1
2016
... 为了进一步探究相稳定、力学及热力学性质产生的物理根源,有必要研究基体元素和添加元素之间的价键作用.但是基于投影缀加波(PAW)交换关联势的密度泛函理论方法得到的电子结构无法直接给出原子之间的价键作用信息,因此本工作进一步计算了投影晶体轨道Hamilton布局(projected crystal orbital Hamiltonian population,pCOHP)[24],对原子轨道电子重叠情况进行分析.pCOHP是一种将密度泛函理论所得到的能带波函数原子轨道化处理的方法,是Deringer等[24]在1983年Hughbanks和Hoffmann[25]提出的晶体轨道重合布局(crystal orbital overlap population,COOP)和1993年Dronskowski和Blöch[26]发展的晶体轨道Hamilton布局(crystal orbital Hamilton population,COHP)分析的基础上进一步发展起来的.在pCOHP方法中,依次对能带波函数进行转移矩阵(包含单电子轨道函数)处理、投影密度矩阵(包含基于原子轨道线性组合获得的相关原子轨道化学信息)处理、Hamilton矩阵化处理,最后将该Hamilton布局权重作用到态密度曲线上,所得即pCOHP曲线[24].曲线包含成键和反键轨道电子信息,成键轨道电子引起能量下降,而反键轨道电子引起能量增加,可以定量表征键强,适用于共价键、离子键、金属键以及分子键体系[24].本工作中,pCOHP曲线基于面向电子结构重建的局域轨道基组(local orbital suite towards electronic-structure reconstruction,LOBSTER)程序[27]计算获得.除此之外,本工作利用二次差分电荷密度来分析掺杂前后电子的转移以及空间分布情况. ...
Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set
1
1996
... 计算利用VASP软件包[28],基于密度泛函理论展开.离子实和电子之间的相互作用通过PAW描述[29,30].使用应力收敛小于10-2 eV/nm的共轭梯度算法将应力和Hellman-Feynman力最小化以确定基态几何结构.使用450 eV的平面截断能来扩展Kohn-Sham轨道.对于结构优化和总能量计算,在Brillouin区积分上,W-Ir结构使用4 × 4 × 4 Monkhorst-Pack K点网格[31,32],W和Ir的价电子构型分别为6s5d4、6s5d7. ...
Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set
1
1996
... 计算利用VASP软件包[28],基于密度泛函理论展开.离子实和电子之间的相互作用通过PAW描述[29,30].使用应力收敛小于10-2 eV/nm的共轭梯度算法将应力和Hellman-Feynman力最小化以确定基态几何结构.使用450 eV的平面截断能来扩展Kohn-Sham轨道.对于结构优化和总能量计算,在Brillouin区积分上,W-Ir结构使用4 × 4 × 4 Monkhorst-Pack K点网格[31,32],W和Ir的价电子构型分别为6s5d4、6s5d7. ...
Generalized gradient approximation made simple
1
1996
... 计算利用VASP软件包[28],基于密度泛函理论展开.离子实和电子之间的相互作用通过PAW描述[29,30].使用应力收敛小于10-2 eV/nm的共轭梯度算法将应力和Hellman-Feynman力最小化以确定基态几何结构.使用450 eV的平面截断能来扩展Kohn-Sham轨道.对于结构优化和总能量计算,在Brillouin区积分上,W-Ir结构使用4 × 4 × 4 Monkhorst-Pack K点网格[31,32],W和Ir的价电子构型分别为6s5d4、6s5d7. ...
Projector augmented-wave method
1
1994
... 计算利用VASP软件包[28],基于密度泛函理论展开.离子实和电子之间的相互作用通过PAW描述[29,30].使用应力收敛小于10-2 eV/nm的共轭梯度算法将应力和Hellman-Feynman力最小化以确定基态几何结构.使用450 eV的平面截断能来扩展Kohn-Sham轨道.对于结构优化和总能量计算,在Brillouin区积分上,W-Ir结构使用4 × 4 × 4 Monkhorst-Pack K点网格[31,32],W和Ir的价电子构型分别为6s5d4、6s5d7. ...
Special points for Brillouin-zone integrations
1
1976
... 计算利用VASP软件包[28],基于密度泛函理论展开.离子实和电子之间的相互作用通过PAW描述[29,30].使用应力收敛小于10-2 eV/nm的共轭梯度算法将应力和Hellman-Feynman力最小化以确定基态几何结构.使用450 eV的平面截断能来扩展Kohn-Sham轨道.对于结构优化和总能量计算,在Brillouin区积分上,W-Ir结构使用4 × 4 × 4 Monkhorst-Pack K点网格[31,32],W和Ir的价电子构型分别为6s5d4、6s5d7. ...
Results of the IUCr precision lattice-parameter project
3
1960
... 为了验证计算方法以及所用参数的可靠性,首先计算了纯金属W和Ir的晶格常数和弹性模量.计算结果以及前人已发表工作[33~48]的对比示于表1.可以看到,当前计算所得晶格常数、弹性模量与其他实验以及计算所得值符合很好.参量G / B和Cp均预测出了W和Ir的脆性特征,计算所得值也在其他实验和计算值范围之内.由此可见,当前的计算方法和所用参数在预测合金对W基体的晶体结构、弹性力学性能方面是合理和可靠的. ...
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... 表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
Equations of state of six metals above 94 GPa
1
2004
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
Elastic constants of tantalum, tungsten, and molybdenum
7
1963
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
35], 164
[36]418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] | | Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
35], 419
[36]0.52[35], 0.52[36] | 41.82[35], 41.79[36] | | Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
35], 0.52
[36]41.82[35], 41.79[36] | | Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
35], 41.79
[36] | Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... W-Ir二元合金和纯W[35~41]的弹性常数(C11、C12、C44)、弹性模量(B、G、E)、G / B和Cp随Ir添加量的变化曲线示于图4.可以看到,纯W计算值均在其他实验和计算所得结果范围之内,表明了当前弹性系数计算方法和参数设置的可靠性.弹性系数C11表征了晶体材料在承受单轴应变时的抗变形能力,C12和C44则分别表征了晶体材料沿着(00l) (l为晶面指数)侧面和<111>四度轴向的抗剪切能力.图4表明,随着Ir含量的增加,C11和C44呈减小趋势,表明Ir的加入可以明显降低单轴受力和<111>轴向剪切时的抗变形能力,提升材料韧性;而C12则随Ir含量增加而单调增加,表明合金抗平面剪切变形能力增加,预示该方向变形时材料刚度增加;弹性模量B、G和E均随着Ir的添加单调递减,表明Ir的添加会降低材料发生体积变形、剪切变形和单轴变形时的抗变形能力.这一结果预示W-Ir合金在遭受外部应力时,W-Ir合金体系比纯W体系更易达到剪切应力极限诱发位错发射,通过塑性协变缓解裂纹尖端应力集中,稳定了裂纹尖端(韧性特征),避免或者延缓了脆性断裂.这种Ir诱发的易变形特性也从另一个角度验证了图2中形成能的计算结果,即Ir能诱发相的不稳定特性.随着Ir含量从0增加到12.96%,表征韧脆特征的参量G / B从0.52 (脆性)降低到0.37 (韧性);Cp为正值,且随着Ir含量的增加从42 GPa增加到112 GPa,也预示着Ir的添加能改善W基体的脆性,提升其韧性[18].这一结论与Luo等[8,9]和McKamey等[10]的实验观察一致.该实验研究发现,Ir的加入会导致钨合金的极限拉伸应力和屈服强度下降,韧性增加.另外,对比Ir和Mo在W中的韧化效果,Mo含量从0变化至70%,G / B仅降低0.03[49],而Ir含量从0变化到12.96%,G / B降低0.15,可见Ir对W的韧化效果远远大于Mo对W的韧化作用.本工作将进一步从电子价键的角度去分析其韧化机理. ...
... [
35~
41]的弹性常数(
C11、C12、C44)、弹性模量(
B、G、E)、
G /
B和
Cp随Ir添加量的变化曲线
Variations of elastic constants (<i>C</i><sub>11</sub>, <i>C</i><sub>12</sub>, <i>C</i><sub>44</sub>) (a), elastic moduli (<i>B</i>, <i>G</i>, <i>E</i>) (b), <i>G</i> / <i>B</i> (c), and <i>C</i><sub>p</sub> (d) with Ir additions for W-Ir binary alloys (The corresponding data of bcc-W from literatures [35-41]<sup/>are also presented for comparison)Fig.4![]()
虽然整体上Ir对W的合金化作用呈现韧性,但是W-Ir合金的平面剪切强化特性不可忽略,需慎重考虑特殊服役环境下的应力发展情况.尤其是当晶体结构具有大量明显的特征取向(如织构)时,要尽量避免外力加载方向与(00l)方向平行,此时W-Ir合金易发生脆性形变. ...
Elastic constants of tungsten between 4.2 and 77 K
5
1980
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
36]
418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] | | Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
36]
0.52[35], 0.52[36] | 41.82[35], 41.79[36] | | Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
36]
41.82[35], 41.79[36] | | Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
36]
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
Theory of elastic constants of cubic transition metals and alloys
6
1993
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
37], 310
[38],
176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], | | | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
37], 145
[38],
447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], | | | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
37], 377
[38],
0.55[37], 0.47[38], | 29[37], 66.70[38], | | | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
37], 0.47
[38],
29[37], 66.70[38], | | | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
37], 66.70
[38],
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
First-principles equations of state and elastic properties of seven metals
5
2005
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
38],
447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], | | | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
38],
0.55[37], 0.47[38], | 29[37], 66.70[38], | | | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
38],
29[37], 66.70[38], | | | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
38],
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
Elasticity of the superconducting metals V, Nb, Ta, Mo, and W at high pressure
3
2008
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
39], 148
[40]379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] | | Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
39], 382
[40]0.49[40], 0.48[41] | 52.7[40], 59[41] | | Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
First-principles calculations of elastic, phonon and thermodynamic properties of W
5
2016
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
40]
379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] | | Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
40]
0.49[40], 0.48[41] | 52.7[40], 59[41] | | Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
40], 0.48
[41]52.7[40], 59[41] | | Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
40], 59
[41]| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
Elastic constants of single‐crystal Mo and W between 77° and 500°K
4
1962
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
41]
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... W-Ir二元合金和纯W[35~41]的弹性常数(C11、C12、C44)、弹性模量(B、G、E)、G / B和Cp随Ir添加量的变化曲线示于图4.可以看到,纯W计算值均在其他实验和计算所得结果范围之内,表明了当前弹性系数计算方法和参数设置的可靠性.弹性系数C11表征了晶体材料在承受单轴应变时的抗变形能力,C12和C44则分别表征了晶体材料沿着(00l) (l为晶面指数)侧面和<111>四度轴向的抗剪切能力.图4表明,随着Ir含量的增加,C11和C44呈减小趋势,表明Ir的加入可以明显降低单轴受力和<111>轴向剪切时的抗变形能力,提升材料韧性;而C12则随Ir含量增加而单调增加,表明合金抗平面剪切变形能力增加,预示该方向变形时材料刚度增加;弹性模量B、G和E均随着Ir的添加单调递减,表明Ir的添加会降低材料发生体积变形、剪切变形和单轴变形时的抗变形能力.这一结果预示W-Ir合金在遭受外部应力时,W-Ir合金体系比纯W体系更易达到剪切应力极限诱发位错发射,通过塑性协变缓解裂纹尖端应力集中,稳定了裂纹尖端(韧性特征),避免或者延缓了脆性断裂.这种Ir诱发的易变形特性也从另一个角度验证了图2中形成能的计算结果,即Ir能诱发相的不稳定特性.随着Ir含量从0增加到12.96%,表征韧脆特征的参量G / B从0.52 (脆性)降低到0.37 (韧性);Cp为正值,且随着Ir含量的增加从42 GPa增加到112 GPa,也预示着Ir的添加能改善W基体的脆性,提升其韧性[18].这一结论与Luo等[8,9]和McKamey等[10]的实验观察一致.该实验研究发现,Ir的加入会导致钨合金的极限拉伸应力和屈服强度下降,韧性增加.另外,对比Ir和Mo在W中的韧化效果,Mo含量从0变化至70%,G / B仅降低0.03[49],而Ir含量从0变化到12.96%,G / B降低0.15,可见Ir对W的韧化效果远远大于Mo对W的韧化作用.本工作将进一步从电子价键的角度去分析其韧化机理. ...
... ~
41]的弹性常数(
C11、C12、C44)、弹性模量(
B、G、E)、
G /
B和
Cp随Ir添加量的变化曲线
Variations of elastic constants (<i>C</i><sub>11</sub>, <i>C</i><sub>12</sub>, <i>C</i><sub>44</sub>) (a), elastic moduli (<i>B</i>, <i>G</i>, <i>E</i>) (b), <i>G</i> / <i>B</i> (c), and <i>C</i><sub>p</sub> (d) with Ir additions for W-Ir binary alloys (The corresponding data of bcc-W from literatures [35-41]<sup/>are also presented for comparison)Fig.4![]()
虽然整体上Ir对W的合金化作用呈现韧性,但是W-Ir合金的平面剪切强化特性不可忽略,需慎重考虑特殊服役环境下的应力发展情况.尤其是当晶体结构具有大量明显的特征取向(如织构)时,要尽量避免外力加载方向与(00l)方向平行,此时W-Ir合金易发生脆性形变. ...
Ir-base refractory superalloys for ultra-high temperatures
1
1998
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
5
1971
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
43], 217
[44]550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] | | Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
43], 540
[44]0.61[43], 0.61[44] | -13[43], -14[44] | | Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
43], 0.61
[44]-13[43], -14[44] | | Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
43], -14
[44] | Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
5
2005
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
44]
550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] | | Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
44]
0.61[43], 0.61[44] | -13[43], -14[44] | | Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
44]
-13[43], -14[44] | | Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
44]
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
Modeling the elastic anisotropies and mechanical strengths of Ir3 X intermetallics
6
2017
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
45], 364
[46],
232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], | | | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
45], 223
[46],
570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], | | | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
45], 555
[46],
0.66[45], 0.61[46], | -43[45], -15[46], | | | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
45], 0.61
[46],
-43[45], -15[46], | | | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
45], -15
[46],
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
Iridium concentration driving the mechanical properties of iridium-aluminum compounds
5
2015
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
46],
570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], | | | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
46],
0.66[45], 0.61[46], | -43[45], -15[46], | | | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
46],
-43[45], -15[46], | | | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
46],
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
Applications of a tight-binding total-energy method for transition and noble metals: Elastic constants, vacancies, and surfaces of monatomic metals
5
1996
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
47], 222
[48]698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] | 表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
47], 549
[48]0.71[47], 0.64[48] | -88[47], -29[48] | 表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
47], 0.64
[48]-88[47], -29[48] | 表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
47], -29
[48]表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
Ab initio examination of ductility features of fcc metals
7
2009
... 为了验证计算方法以及所用参数的可靠性,首先计算了纯金属W和Ir的晶格常数和弹性模量.计算结果以及前人已发表工作[33~48]的对比示于表1.可以看到,当前计算所得晶格常数、弹性模量与其他实验以及计算所得值符合很好.参量G / B和Cp均预测出了W和Ir的脆性特征,计算所得值也在其他实验和计算值范围之内.由此可见,当前的计算方法和所用参数在预测合金对W基体的晶体结构、弹性力学性能方面是合理和可靠的. ...
... Comparisons between the calculated lattice constant (
a), elastic moduli (bulk modulus
(
B), shear modulus (
G) and Young's modulus
(
E)), Pugh value (
G /
B) and Cauchy pressure (
Cp) of W and Ir according to Equations (6)-(8) and those from the literatures [33-48]
| Element | Source | a / nm | B / GPa | G / GPa | E / GPa | G / B | Cp / GPa |
|---|
| W | Present | 0.3171 | 305 | 158 | 404 | 0.52 | 41.71 |
| Exp. | 0.3165[33], 0.3166[34] | 314[35], 315[36] | 163[35], 164[36] | 418[35], 419[36] | 0.52[35], 0.52[36] | 41.82[35], 41.79[36] |
| Calc. | 0.3171[37] | 323[37], 310[38], | 176[37], 145[38], | 447[37], 377[38], | 0.55[37], 0.47[38], | 29[37], 66.70[38], |
| | | 304[39], 301[40] | 147[39], 148[40] | 379[39], 382[40] | 0.49[40], 0.48[41] | 52.7[40], 59[41] |
| Ir | Present | 0.3877 | 342 | 226 | 555 | 0.66 | -39.07 |
| Exp. | 0.3839[42] | 363[43], 353[44] | 221[43], 217[44] | 550[43], 540[44] | 0.61[43], 0.61[44] | -13[43], -14[44] |
| Calc. | 0.3871[45] | 351[45], 364[46], | 232[45], 223[46], | 570[45], 555[46], | 0.66[45], 0.61[46], | -43[45], -15[46], |
| | | 405[47], 347[48] | 288[47], 222[48] | 698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] |
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
48]
698[47], 549[48] | 0.71[47], 0.64[48] | -88[47], -29[48] | 表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
48]
0.71[47], 0.64[48] | -88[47], -29[48] | 表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
48]
-88[47], -29[48] | 表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... [
48]
表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
... 表1 计算所得W和Ir的晶格常数(a)、弹性模量(体积弹性模量(B)、剪切模量(G)和Young's模量(E))、Pugh值(G / B)和Cauchy应力(Cp)与文献[33~48]中弹性常数按式(6)~(8)计算所得数据的比较 ...
A first principles investigation of W1 - x Mo x (x = 0-68.75 at.%) alloys: Structural, electronic, mechanical and thermal properties
1
2020
... W-Ir二元合金和纯W[35~41]的弹性常数(C11、C12、C44)、弹性模量(B、G、E)、G / B和Cp随Ir添加量的变化曲线示于图4.可以看到,纯W计算值均在其他实验和计算所得结果范围之内,表明了当前弹性系数计算方法和参数设置的可靠性.弹性系数C11表征了晶体材料在承受单轴应变时的抗变形能力,C12和C44则分别表征了晶体材料沿着(00l) (l为晶面指数)侧面和<111>四度轴向的抗剪切能力.图4表明,随着Ir含量的增加,C11和C44呈减小趋势,表明Ir的加入可以明显降低单轴受力和<111>轴向剪切时的抗变形能力,提升材料韧性;而C12则随Ir含量增加而单调增加,表明合金抗平面剪切变形能力增加,预示该方向变形时材料刚度增加;弹性模量B、G和E均随着Ir的添加单调递减,表明Ir的添加会降低材料发生体积变形、剪切变形和单轴变形时的抗变形能力.这一结果预示W-Ir合金在遭受外部应力时,W-Ir合金体系比纯W体系更易达到剪切应力极限诱发位错发射,通过塑性协变缓解裂纹尖端应力集中,稳定了裂纹尖端(韧性特征),避免或者延缓了脆性断裂.这种Ir诱发的易变形特性也从另一个角度验证了图2中形成能的计算结果,即Ir能诱发相的不稳定特性.随着Ir含量从0增加到12.96%,表征韧脆特征的参量G / B从0.52 (脆性)降低到0.37 (韧性);Cp为正值,且随着Ir含量的增加从42 GPa增加到112 GPa,也预示着Ir的添加能改善W基体的脆性,提升其韧性[18].这一结论与Luo等[8,9]和McKamey等[10]的实验观察一致.该实验研究发现,Ir的加入会导致钨合金的极限拉伸应力和屈服强度下降,韧性增加.另外,对比Ir和Mo在W中的韧化效果,Mo含量从0变化至70%,G / B仅降低0.03[49],而Ir含量从0变化到12.96%,G / B降低0.15,可见Ir对W的韧化效果远远大于Mo对W的韧化作用.本工作将进一步从电子价键的角度去分析其韧化机理. ...
1
1988
... 由图6差分电荷密度差值的形状和分布,参照金属(合金)轨道电荷分布拓扑图以及不同轨道的能量分布[50],W-Ir合金中W和Ir的电荷密度应该是按照如下方式重新分布为:Ir原子中能量较高的轨道失去电子(图6中蓝色区域),转移并填充到能量较低的d xz 和d yz 轨道;而W中的电子则是从能量较低的轨道转移到能量较高的d xz 和d yz 轨道,Ir与W之间通过d xz 和d yz 轨道重叠作用而成键,相应轨道电荷增强(图6中红色和黄色区域).这种键合方式虽然略微降低了W的稳定性,但是外层d电子数较多的Ir对W的韧化作用非常显著. ...



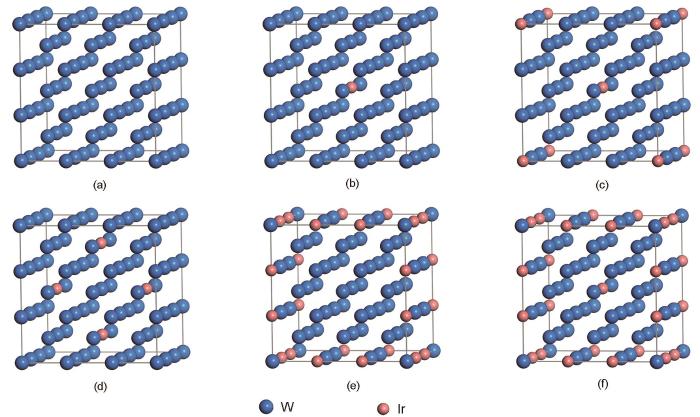
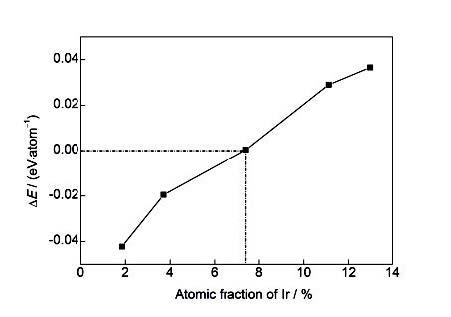

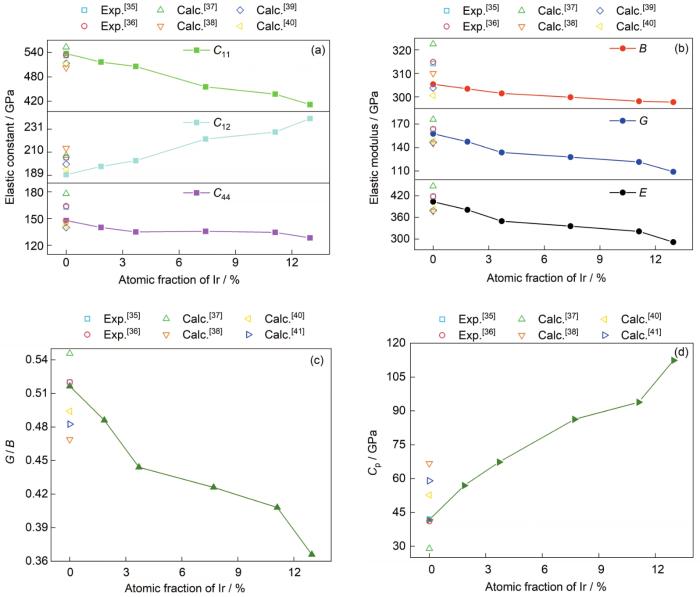
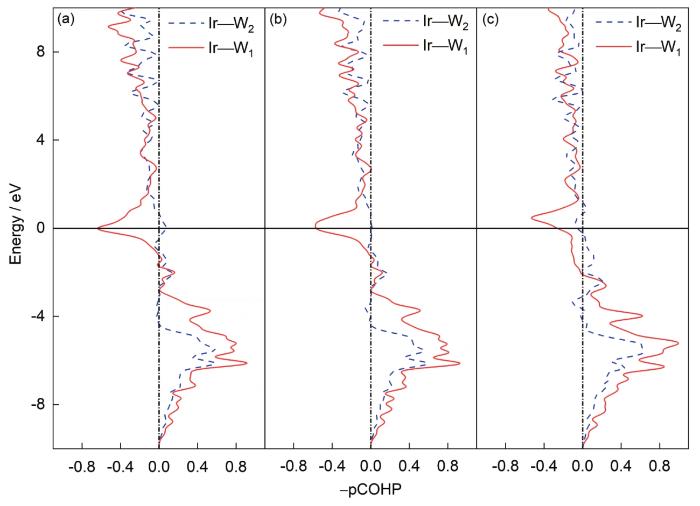
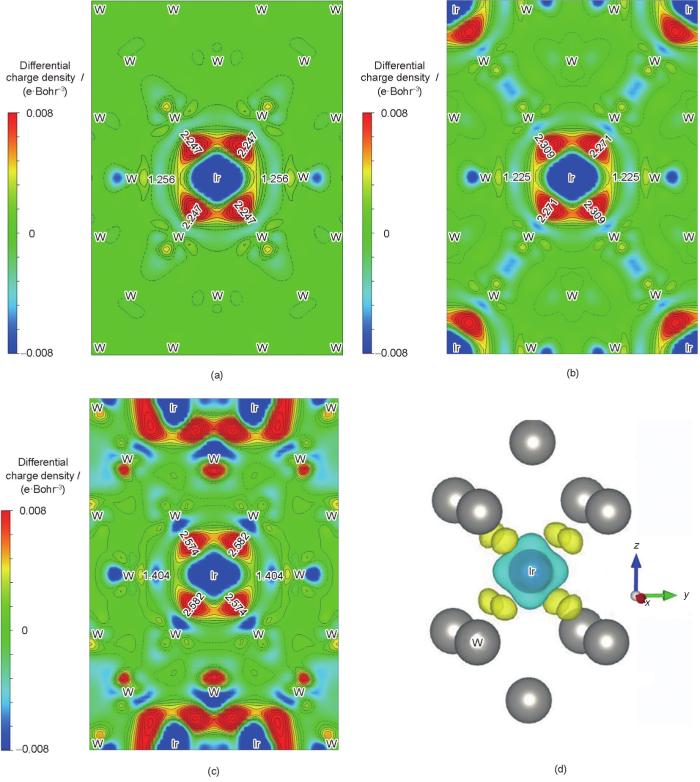
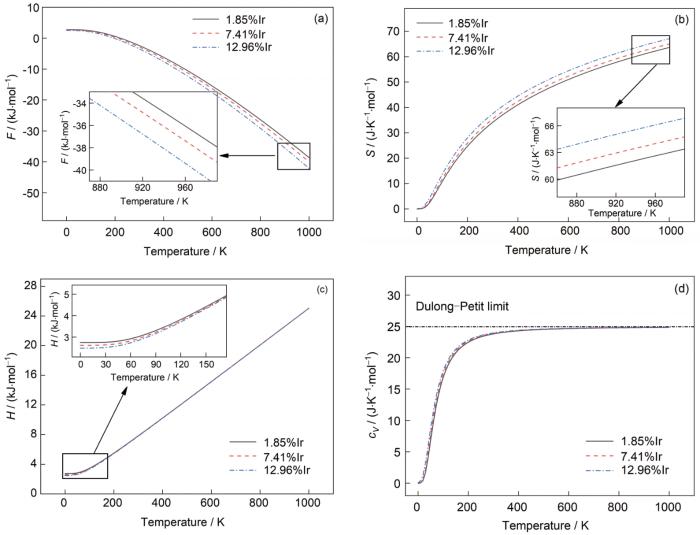

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}