Deep potential molecular dynamics: A scalable model with the accuracy of quantum mechanics
2
2018
... 近年来,在AI for Science (AI4S)理念的引领下,科学领域迎来了许多与人工智能相关的前沿进展.例如,基于深度势能的分子动力学 (deep potential for molecular dynamics,DeePMD)方法[1,2]能在量子力学精度模拟上亿原子,如今已成为原子尺度模拟的重要工具.在基于机器学习的势函数方法里,需要通过密度泛函理论(density functional theory,DFT)[3,4]软件等第一性原理计算软件算出体系的总能量、原子受力和晶胞所受应力等物理性质,然后把原子构型和对应的这些物理性质作为机器学习模型的“标签”,从而训练出具有第一性原理精度的深度神经网络势函数. ...
... 基于深度学习的原子间势函数方法例如深度势能(deep potential)方法[1,2]是一种典型的监督学习方法,通过训练第一性原理数据产生原子间势函数模型,目前已经被广泛应用于各种材料计算场景.在该框架下,系统总能量E的表达式可写成体系里n个原子(指标为)的能量之和: ...
DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics
3
2018
... 近年来,在AI for Science (AI4S)理念的引领下,科学领域迎来了许多与人工智能相关的前沿进展.例如,基于深度势能的分子动力学 (deep potential for molecular dynamics,DeePMD)方法[1,2]能在量子力学精度模拟上亿原子,如今已成为原子尺度模拟的重要工具.在基于机器学习的势函数方法里,需要通过密度泛函理论(density functional theory,DFT)[3,4]软件等第一性原理计算软件算出体系的总能量、原子受力和晶胞所受应力等物理性质,然后把原子构型和对应的这些物理性质作为机器学习模型的“标签”,从而训练出具有第一性原理精度的深度神经网络势函数. ...
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
... 基于深度学习的原子间势函数方法例如深度势能(deep potential)方法[1,2]是一种典型的监督学习方法,通过训练第一性原理数据产生原子间势函数模型,目前已经被广泛应用于各种材料计算场景.在该框架下,系统总能量E的表达式可写成体系里n个原子(指标为)的能量之和: ...
Inhomogeneous electron gas
2
1964
... 近年来,在AI for Science (AI4S)理念的引领下,科学领域迎来了许多与人工智能相关的前沿进展.例如,基于深度势能的分子动力学 (deep potential for molecular dynamics,DeePMD)方法[1,2]能在量子力学精度模拟上亿原子,如今已成为原子尺度模拟的重要工具.在基于机器学习的势函数方法里,需要通过密度泛函理论(density functional theory,DFT)[3,4]软件等第一性原理计算软件算出体系的总能量、原子受力和晶胞所受应力等物理性质,然后把原子构型和对应的这些物理性质作为机器学习模型的“标签”,从而训练出具有第一性原理精度的深度神经网络势函数. ...
... 密度泛函理论基于Hohenberg-Kohn定理[3],该定理将体系的电子密度作为基本变量并将体系的总能量定义为电子密度的泛函.其中,KSDFT在精度和计算效率间取得了较好平衡,Kohn-Sham方程是目前主流使用的DFT方法,其求解过程如下[4]: ...
Self-consistent equations including exchange and correlation effects
2
1965
... 近年来,在AI for Science (AI4S)理念的引领下,科学领域迎来了许多与人工智能相关的前沿进展.例如,基于深度势能的分子动力学 (deep potential for molecular dynamics,DeePMD)方法[1,2]能在量子力学精度模拟上亿原子,如今已成为原子尺度模拟的重要工具.在基于机器学习的势函数方法里,需要通过密度泛函理论(density functional theory,DFT)[3,4]软件等第一性原理计算软件算出体系的总能量、原子受力和晶胞所受应力等物理性质,然后把原子构型和对应的这些物理性质作为机器学习模型的“标签”,从而训练出具有第一性原理精度的深度神经网络势函数. ...
... 密度泛函理论基于Hohenberg-Kohn定理[3],该定理将体系的电子密度作为基本变量并将体系的总能量定义为电子密度的泛函.其中,KSDFT在精度和计算效率间取得了较好平衡,Kohn-Sham方程是目前主流使用的DFT方法,其求解过程如下[4]: ...
Roadmap on machine learning in electronic structure
1
2022
... 近年来,基于机器学习的DFT方法处于快速发展阶段[5].例如,Chen等[6]提出了deep Kohn-Sham (DeePKS)方法,基于局域基组构造电子结构的描述子,通过在电子体系的Hamiltonian量里添加基于深度学习的泛函修正项,学习低精度泛函和高精度泛函所预测的性质之差,从而较好地平衡效率和精度.2021年DeepMind公司的研究人员基于PySCF软件发布了基于深度学习的神经网络模型——DM21泛函[7],该泛函的精度在一些给定测试体系上高于杂化泛函(hybrid functional)甚至接近双杂化泛函.Li等[8~10]提出基于深度学习和局域基组的Hamiltonian矩阵构建(deep-learing DFT Hamiltonian,DeepH)方法以及基于深度学习的密度泛函微扰理论(density functional perturbation theory,DFPT),提高了二维材料等体系的电子结构计算效率.来自美国Sandia国家实验室和来自德国Helmholtz-Zentrum Dresden-Rossendorf研究机构的科研人员发起了materials learning algorithms (MALA)项目,该研究旨在通过深度学习的方法加速DFT计算,目前已应用于Kohn-Sham密度泛函理论(Kohn-Sham DFT,KSDFT)[11]和无轨道密度泛函理论(orbital-free DFT,OFDFT)[12].Gong等[13]提出通过基于机器学习的对比学习方法可有效地对交换关联泛函进行物理限制,从而提高交换关联泛函精度. ...
DeePKS-kit: A package for developing machine learning-based chemically accurate energy and density functional models
3
2023
... 近年来,基于机器学习的DFT方法处于快速发展阶段[5].例如,Chen等[6]提出了deep Kohn-Sham (DeePKS)方法,基于局域基组构造电子结构的描述子,通过在电子体系的Hamiltonian量里添加基于深度学习的泛函修正项,学习低精度泛函和高精度泛函所预测的性质之差,从而较好地平衡效率和精度.2021年DeepMind公司的研究人员基于PySCF软件发布了基于深度学习的神经网络模型——DM21泛函[7],该泛函的精度在一些给定测试体系上高于杂化泛函(hybrid functional)甚至接近双杂化泛函.Li等[8~10]提出基于深度学习和局域基组的Hamiltonian矩阵构建(deep-learing DFT Hamiltonian,DeepH)方法以及基于深度学习的密度泛函微扰理论(density functional perturbation theory,DFPT),提高了二维材料等体系的电子结构计算效率.来自美国Sandia国家实验室和来自德国Helmholtz-Zentrum Dresden-Rossendorf研究机构的科研人员发起了materials learning algorithms (MALA)项目,该研究旨在通过深度学习的方法加速DFT计算,目前已应用于Kohn-Sham密度泛函理论(Kohn-Sham DFT,KSDFT)[11]和无轨道密度泛函理论(orbital-free DFT,OFDFT)[12].Gong等[13]提出通过基于机器学习的对比学习方法可有效地对交换关联泛函进行物理限制,从而提高交换关联泛函精度. ...
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
... 机器学习势函数方法的瓶颈之一是在势函数的训练过程中需要生成大量的第一性原理计算数据,这些计算不仅需要较大的成本,还使得机器学习势函数的生成需要较长的周期,降低了项目快速迭代的可能性.若需使用高精度(例如杂化泛函)的第一性原理方法来产生数据,则会极大增加机器学习势函数的训练数据成本,从而限制了机器学习势函数的应用范围.针对此类问题,采用基于机器学习的DeePKS方法[70]以及相应的DeePKS-kit软件[6]可以有效解决精度和效率难两全的困境. ...
Pushing the frontiers of density functionals by solving the fractional electron problem
1
2021
... 近年来,基于机器学习的DFT方法处于快速发展阶段[5].例如,Chen等[6]提出了deep Kohn-Sham (DeePKS)方法,基于局域基组构造电子结构的描述子,通过在电子体系的Hamiltonian量里添加基于深度学习的泛函修正项,学习低精度泛函和高精度泛函所预测的性质之差,从而较好地平衡效率和精度.2021年DeepMind公司的研究人员基于PySCF软件发布了基于深度学习的神经网络模型——DM21泛函[7],该泛函的精度在一些给定测试体系上高于杂化泛函(hybrid functional)甚至接近双杂化泛函.Li等[8~10]提出基于深度学习和局域基组的Hamiltonian矩阵构建(deep-learing DFT Hamiltonian,DeepH)方法以及基于深度学习的密度泛函微扰理论(density functional perturbation theory,DFPT),提高了二维材料等体系的电子结构计算效率.来自美国Sandia国家实验室和来自德国Helmholtz-Zentrum Dresden-Rossendorf研究机构的科研人员发起了materials learning algorithms (MALA)项目,该研究旨在通过深度学习的方法加速DFT计算,目前已应用于Kohn-Sham密度泛函理论(Kohn-Sham DFT,KSDFT)[11]和无轨道密度泛函理论(orbital-free DFT,OFDFT)[12].Gong等[13]提出通过基于机器学习的对比学习方法可有效地对交换关联泛函进行物理限制,从而提高交换关联泛函精度. ...
Deep-learning density functional perturbation theory
3
2024
... 近年来,基于机器学习的DFT方法处于快速发展阶段[5].例如,Chen等[6]提出了deep Kohn-Sham (DeePKS)方法,基于局域基组构造电子结构的描述子,通过在电子体系的Hamiltonian量里添加基于深度学习的泛函修正项,学习低精度泛函和高精度泛函所预测的性质之差,从而较好地平衡效率和精度.2021年DeepMind公司的研究人员基于PySCF软件发布了基于深度学习的神经网络模型——DM21泛函[7],该泛函的精度在一些给定测试体系上高于杂化泛函(hybrid functional)甚至接近双杂化泛函.Li等[8~10]提出基于深度学习和局域基组的Hamiltonian矩阵构建(deep-learing DFT Hamiltonian,DeepH)方法以及基于深度学习的密度泛函微扰理论(density functional perturbation theory,DFPT),提高了二维材料等体系的电子结构计算效率.来自美国Sandia国家实验室和来自德国Helmholtz-Zentrum Dresden-Rossendorf研究机构的科研人员发起了materials learning algorithms (MALA)项目,该研究旨在通过深度学习的方法加速DFT计算,目前已应用于Kohn-Sham密度泛函理论(Kohn-Sham DFT,KSDFT)[11]和无轨道密度泛函理论(orbital-free DFT,OFDFT)[12].Gong等[13]提出通过基于机器学习的对比学习方法可有效地对交换关联泛函进行物理限制,从而提高交换关联泛函精度. ...
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
... 此外ABACUS 3.0版本还支持与其他AI算法结合,例如支持主动学习生成机器学习势函数的DP-GEN[74]方法,以及支持基于原子位置生成Hamiltonian矩阵的DeepH[8]方法,这些新算法可以进一步提升电子和原子尺度的计算精度和效率. ...
Deep-learning density functional theory Hamiltonian for efficient ab initio electronic-structure calculation
0
2022
General framework for E(3)-equivariant neural network representation of density functional theory Hamiltonian
1
2023
... 近年来,基于机器学习的DFT方法处于快速发展阶段[5].例如,Chen等[6]提出了deep Kohn-Sham (DeePKS)方法,基于局域基组构造电子结构的描述子,通过在电子体系的Hamiltonian量里添加基于深度学习的泛函修正项,学习低精度泛函和高精度泛函所预测的性质之差,从而较好地平衡效率和精度.2021年DeepMind公司的研究人员基于PySCF软件发布了基于深度学习的神经网络模型——DM21泛函[7],该泛函的精度在一些给定测试体系上高于杂化泛函(hybrid functional)甚至接近双杂化泛函.Li等[8~10]提出基于深度学习和局域基组的Hamiltonian矩阵构建(deep-learing DFT Hamiltonian,DeepH)方法以及基于深度学习的密度泛函微扰理论(density functional perturbation theory,DFPT),提高了二维材料等体系的电子结构计算效率.来自美国Sandia国家实验室和来自德国Helmholtz-Zentrum Dresden-Rossendorf研究机构的科研人员发起了materials learning algorithms (MALA)项目,该研究旨在通过深度学习的方法加速DFT计算,目前已应用于Kohn-Sham密度泛函理论(Kohn-Sham DFT,KSDFT)[11]和无轨道密度泛函理论(orbital-free DFT,OFDFT)[12].Gong等[13]提出通过基于机器学习的对比学习方法可有效地对交换关联泛函进行物理限制,从而提高交换关联泛函精度. ...
Accelerating finite-temperature Kohn-Sham density functional theory with deep neural networks
1
2021
... 近年来,基于机器学习的DFT方法处于快速发展阶段[5].例如,Chen等[6]提出了deep Kohn-Sham (DeePKS)方法,基于局域基组构造电子结构的描述子,通过在电子体系的Hamiltonian量里添加基于深度学习的泛函修正项,学习低精度泛函和高精度泛函所预测的性质之差,从而较好地平衡效率和精度.2021年DeepMind公司的研究人员基于PySCF软件发布了基于深度学习的神经网络模型——DM21泛函[7],该泛函的精度在一些给定测试体系上高于杂化泛函(hybrid functional)甚至接近双杂化泛函.Li等[8~10]提出基于深度学习和局域基组的Hamiltonian矩阵构建(deep-learing DFT Hamiltonian,DeepH)方法以及基于深度学习的密度泛函微扰理论(density functional perturbation theory,DFPT),提高了二维材料等体系的电子结构计算效率.来自美国Sandia国家实验室和来自德国Helmholtz-Zentrum Dresden-Rossendorf研究机构的科研人员发起了materials learning algorithms (MALA)项目,该研究旨在通过深度学习的方法加速DFT计算,目前已应用于Kohn-Sham密度泛函理论(Kohn-Sham DFT,KSDFT)[11]和无轨道密度泛函理论(orbital-free DFT,OFDFT)[12].Gong等[13]提出通过基于机器学习的对比学习方法可有效地对交换关联泛函进行物理限制,从而提高交换关联泛函精度. ...
Accelerating equilibration in first-principles molecular dynamics with orbital-free density functional theory
1
2022
... 近年来,基于机器学习的DFT方法处于快速发展阶段[5].例如,Chen等[6]提出了deep Kohn-Sham (DeePKS)方法,基于局域基组构造电子结构的描述子,通过在电子体系的Hamiltonian量里添加基于深度学习的泛函修正项,学习低精度泛函和高精度泛函所预测的性质之差,从而较好地平衡效率和精度.2021年DeepMind公司的研究人员基于PySCF软件发布了基于深度学习的神经网络模型——DM21泛函[7],该泛函的精度在一些给定测试体系上高于杂化泛函(hybrid functional)甚至接近双杂化泛函.Li等[8~10]提出基于深度学习和局域基组的Hamiltonian矩阵构建(deep-learing DFT Hamiltonian,DeepH)方法以及基于深度学习的密度泛函微扰理论(density functional perturbation theory,DFPT),提高了二维材料等体系的电子结构计算效率.来自美国Sandia国家实验室和来自德国Helmholtz-Zentrum Dresden-Rossendorf研究机构的科研人员发起了materials learning algorithms (MALA)项目,该研究旨在通过深度学习的方法加速DFT计算,目前已应用于Kohn-Sham密度泛函理论(Kohn-Sham DFT,KSDFT)[11]和无轨道密度泛函理论(orbital-free DFT,OFDFT)[12].Gong等[13]提出通过基于机器学习的对比学习方法可有效地对交换关联泛函进行物理限制,从而提高交换关联泛函精度. ...
Incorporation of density scaling constraint in density functional design via contrastive representation learning
1
2023
... 近年来,基于机器学习的DFT方法处于快速发展阶段[5].例如,Chen等[6]提出了deep Kohn-Sham (DeePKS)方法,基于局域基组构造电子结构的描述子,通过在电子体系的Hamiltonian量里添加基于深度学习的泛函修正项,学习低精度泛函和高精度泛函所预测的性质之差,从而较好地平衡效率和精度.2021年DeepMind公司的研究人员基于PySCF软件发布了基于深度学习的神经网络模型——DM21泛函[7],该泛函的精度在一些给定测试体系上高于杂化泛函(hybrid functional)甚至接近双杂化泛函.Li等[8~10]提出基于深度学习和局域基组的Hamiltonian矩阵构建(deep-learing DFT Hamiltonian,DeepH)方法以及基于深度学习的密度泛函微扰理论(density functional perturbation theory,DFPT),提高了二维材料等体系的电子结构计算效率.来自美国Sandia国家实验室和来自德国Helmholtz-Zentrum Dresden-Rossendorf研究机构的科研人员发起了materials learning algorithms (MALA)项目,该研究旨在通过深度学习的方法加速DFT计算,目前已应用于Kohn-Sham密度泛函理论(Kohn-Sham DFT,KSDFT)[11]和无轨道密度泛函理论(orbital-free DFT,OFDFT)[12].Gong等[13]提出通过基于机器学习的对比学习方法可有效地对交换关联泛函进行物理限制,从而提高交换关联泛函精度. ...
Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set
1
1996
... DFT软件是一系列复杂电子结构算法的载体,也是材料计算领域的重要科研工具.近三十年来,国外出现了一批优秀的第一性原理材料计算软件,例如VASP[14,15]、Quantum Espresso[16]、CP2K[17]、SIESTA[18]、OpenMX[19]和FHI-aims[20]等,并且许多软件在算法上还保持着算法和性能的持续创新,引领着DFT领域的发展.目前国内DFT方面的研究工作主要是使用软件进行材料计算研究,也有不少课题组开展了第一性原理算法和软件方面的研究工作,但从规模和社区成熟度方面来说与国外尚有差距. ...
Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set
1
1996
... DFT软件是一系列复杂电子结构算法的载体,也是材料计算领域的重要科研工具.近三十年来,国外出现了一批优秀的第一性原理材料计算软件,例如VASP[14,15]、Quantum Espresso[16]、CP2K[17]、SIESTA[18]、OpenMX[19]和FHI-aims[20]等,并且许多软件在算法上还保持着算法和性能的持续创新,引领着DFT领域的发展.目前国内DFT方面的研究工作主要是使用软件进行材料计算研究,也有不少课题组开展了第一性原理算法和软件方面的研究工作,但从规模和社区成熟度方面来说与国外尚有差距. ...
Quantum espresso: A modular and open-source software project for quantum simulations of materials
1
2009
... DFT软件是一系列复杂电子结构算法的载体,也是材料计算领域的重要科研工具.近三十年来,国外出现了一批优秀的第一性原理材料计算软件,例如VASP[14,15]、Quantum Espresso[16]、CP2K[17]、SIESTA[18]、OpenMX[19]和FHI-aims[20]等,并且许多软件在算法上还保持着算法和性能的持续创新,引领着DFT领域的发展.目前国内DFT方面的研究工作主要是使用软件进行材料计算研究,也有不少课题组开展了第一性原理算法和软件方面的研究工作,但从规模和社区成熟度方面来说与国外尚有差距. ...
CP2K: Atomistic simulations of condensed matter systems
1
2014
... DFT软件是一系列复杂电子结构算法的载体,也是材料计算领域的重要科研工具.近三十年来,国外出现了一批优秀的第一性原理材料计算软件,例如VASP[14,15]、Quantum Espresso[16]、CP2K[17]、SIESTA[18]、OpenMX[19]和FHI-aims[20]等,并且许多软件在算法上还保持着算法和性能的持续创新,引领着DFT领域的发展.目前国内DFT方面的研究工作主要是使用软件进行材料计算研究,也有不少课题组开展了第一性原理算法和软件方面的研究工作,但从规模和社区成熟度方面来说与国外尚有差距. ...
The SIESTA method for ab initio order-N materials simulation
1
2002
... DFT软件是一系列复杂电子结构算法的载体,也是材料计算领域的重要科研工具.近三十年来,国外出现了一批优秀的第一性原理材料计算软件,例如VASP[14,15]、Quantum Espresso[16]、CP2K[17]、SIESTA[18]、OpenMX[19]和FHI-aims[20]等,并且许多软件在算法上还保持着算法和性能的持续创新,引领着DFT领域的发展.目前国内DFT方面的研究工作主要是使用软件进行材料计算研究,也有不少课题组开展了第一性原理算法和软件方面的研究工作,但从规模和社区成熟度方面来说与国外尚有差距. ...
Variationally optimized atomic orbitals for large-scale electronic structures
2
2003
... DFT软件是一系列复杂电子结构算法的载体,也是材料计算领域的重要科研工具.近三十年来,国外出现了一批优秀的第一性原理材料计算软件,例如VASP[14,15]、Quantum Espresso[16]、CP2K[17]、SIESTA[18]、OpenMX[19]和FHI-aims[20]等,并且许多软件在算法上还保持着算法和性能的持续创新,引领着DFT领域的发展.目前国内DFT方面的研究工作主要是使用软件进行材料计算研究,也有不少课题组开展了第一性原理算法和软件方面的研究工作,但从规模和社区成熟度方面来说与国外尚有差距. ...
... 构造精度高、可系统提升数量、可移植性好的原子轨道基组颇具挑战.Junquera等[45]提出在一维量子力学方程中加入约束场,从而求解出具有严格截断的数值原子轨道.Ozaki[19]在OpenMX软件中采用变分法优化局域轨道形状.Blum等[20]提出在全电子DFT软件FHI-aims中,可在一个大的局域轨道基组池中挑选最优的局域轨道,从而构建不同精度的基组.Chen等[46,47]提出利用溢出函数(spillage function)构造可以系统提高轨道数量的数值原子轨道,其中每个数值原子轨道的径向部分由一组球Bessel函数展开.通过对半导体、氧化物、金属、团簇等一系列体系的测试,Li等[48]验证了利用溢出函数的方法产生数值原子轨道基组在求解Kohn-Sham方程时相比平面波基组效率更高,同时保持了良好的精度.Zheng等[49]在ABACUS中实现了基于数值原子轨道基组的高精度应力计算,测试表明相比于平面波基组,数值原子轨道基组在计算收敛应力方面更具优势.Lin等[50]提出了一种拟合数值原子轨道基组的新方案,该方案加入了对参考波函数一阶导数的拟合,进一步提升了数值原子基组的精度和效率.值得注意的是,ABACUS中的数值原子轨道需和对应的赝势一起使用,目前ABACUS官网为用户提供了赝势库和对应的数值原子轨道库文件. ...
Ab initio molecular simulations with numeric atom-centered orbitals
2
2009
... DFT软件是一系列复杂电子结构算法的载体,也是材料计算领域的重要科研工具.近三十年来,国外出现了一批优秀的第一性原理材料计算软件,例如VASP[14,15]、Quantum Espresso[16]、CP2K[17]、SIESTA[18]、OpenMX[19]和FHI-aims[20]等,并且许多软件在算法上还保持着算法和性能的持续创新,引领着DFT领域的发展.目前国内DFT方面的研究工作主要是使用软件进行材料计算研究,也有不少课题组开展了第一性原理算法和软件方面的研究工作,但从规模和社区成熟度方面来说与国外尚有差距. ...
... 构造精度高、可系统提升数量、可移植性好的原子轨道基组颇具挑战.Junquera等[45]提出在一维量子力学方程中加入约束场,从而求解出具有严格截断的数值原子轨道.Ozaki[19]在OpenMX软件中采用变分法优化局域轨道形状.Blum等[20]提出在全电子DFT软件FHI-aims中,可在一个大的局域轨道基组池中挑选最优的局域轨道,从而构建不同精度的基组.Chen等[46,47]提出利用溢出函数(spillage function)构造可以系统提高轨道数量的数值原子轨道,其中每个数值原子轨道的径向部分由一组球Bessel函数展开.通过对半导体、氧化物、金属、团簇等一系列体系的测试,Li等[48]验证了利用溢出函数的方法产生数值原子轨道基组在求解Kohn-Sham方程时相比平面波基组效率更高,同时保持了良好的精度.Zheng等[49]在ABACUS中实现了基于数值原子轨道基组的高精度应力计算,测试表明相比于平面波基组,数值原子轨道基组在计算收敛应力方面更具优势.Lin等[50]提出了一种拟合数值原子轨道基组的新方案,该方案加入了对参考波函数一阶导数的拟合,进一步提升了数值原子基组的精度和效率.值得注意的是,ABACUS中的数值原子轨道需和对应的赝势一起使用,目前ABACUS官网为用户提供了赝势库和对应的数值原子轨道库文件. ...
Norm-conserving pseudopotentials
2
1979
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
... 模守恒赝势(norm-conserving pseudopotentials)[21]是一种广泛应用于描述电子-离子相互作用的近似方法,其目的是在保证计算精度的同时降低计算成本.模守恒赝势通过求解单原子Kohn-Sham方程,获得给定交换关联泛函近似下的原子核与价电子相互作用的近似表达,有效减少了求解Kohn-Sham方程所需电子数,尤其是对高Z (Z为原子序数)元素而言,可极大减少由于考虑全部电子而导致的较大计算量.此外,如果赝势里包含了非局域赝势,则需用到单电子Kohn-Sham波函数.模守恒赝势的构造方式有多种,不同赝势所描述的价电子数量可能不同.一般而言,包含价电子数少的赝势会使DFT计算效率更高,但降低了精度.反之,如果包含的价电子数较多,虽然保证了计算的精度但也降低了计算效率.因此,用户在选择使用赝势的时候需对精度和效率做一定的权衡.与projector augmented-wave (PAW)方法[41]相比,模守恒赝势所采用的能量截断值更高,因此PAW在计算效率方面有优势,但基于PAW的电子结构算法的实现过程相比于模守恒赝势更加复杂.目前,ABACUS软件支持多种格式的模守恒赝势,例如SG15模守恒赝势和Dojo模守恒赝势等,以及支持平面波基组下的超软赝势[42]. ...
Self-interaction correction to density-functional approximations for many-electron systems
1
1981
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
Generalized gradient approximation made simple
2
1996
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
... 式中,左边Hamiltonian量里的四项分别代表电子的动能算符、Hartree势、交换关联(exchange-correlation,简称xc)势和外(external,简称ext)势, r 代表空间坐标,n( r )代表电子密度,εi 代表能级,ψi ( r )代表Kohn-Sham单体波函数.在KSDFT的理论框架里需要对交换关联泛函项进行近似处理,而第一性原理的计算精度往往取决于对交换关联泛函近似的好坏程度.除了早期发展的LDA泛函,目前较为常用的交换关联泛函是GGA泛函,例如Perdew-Burke-Ernzerhof (PBE)泛函[23].此外,Meta-GGA泛函(例如strongly constrained and appropriately-normed (SCAN)泛函[24])近年来也获得了较为广泛的应用[38,39].杂化泛函在一些体系性质的预测上可获得更好结果,例如半导体的能隙修正[35,36],但其计算量相比于GGA泛函也会有数量级的增加. ...
Strongly constrained and appropriately normed semilocal density functional
2
2015
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
... 式中,左边Hamiltonian量里的四项分别代表电子的动能算符、Hartree势、交换关联(exchange-correlation,简称xc)势和外(external,简称ext)势, r 代表空间坐标,n( r )代表电子密度,εi 代表能级,ψi ( r )代表Kohn-Sham单体波函数.在KSDFT的理论框架里需要对交换关联泛函项进行近似处理,而第一性原理的计算精度往往取决于对交换关联泛函近似的好坏程度.除了早期发展的LDA泛函,目前较为常用的交换关联泛函是GGA泛函,例如Perdew-Burke-Ernzerhof (PBE)泛函[23].此外,Meta-GGA泛函(例如strongly constrained and appropriately-normed (SCAN)泛函[24])近年来也获得了较为广泛的应用[38,39].杂化泛函在一些体系性质的预测上可获得更好结果,例如半导体的能隙修正[35,36],但其计算量相比于GGA泛函也会有数量级的增加. ...
Rationale for mixing exact exchange with density functional approximations
1
1996
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
Hybrid functionals based on a screened Coulomb potential
1
2003
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
DP-GEN: A concurrent learning platform for the generation of reliable deep learning based potential energy models
1
2020
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
The atomic simulation environment—A Python library for working with atoms
1
2017
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
Implementation strategies in phonopy and phono3py
1
2023
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
Wannier90: A tool for obtaining maximally-localised Wannier functions
1
2008
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
ShengBTE: A solver of the Boltzmann transport equation for phonons
1
2014
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
Ab initio nonadiabatic molecular dynamics investigations on the excited carriers in condensed matter systems
1
2019
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
Accelerating atomic orbital-based electronic structure calculation via pole expansion and selected inversion
1
2013
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
USPEX—Evolutionary crystal structure prediction
1
2006
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
Efficient hybrid density functional calculations for large periodic systems using numerical atomic orbitals
2
2020
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
... 式中,左边Hamiltonian量里的四项分别代表电子的动能算符、Hartree势、交换关联(exchange-correlation,简称xc)势和外(external,简称ext)势, r 代表空间坐标,n( r )代表电子密度,εi 代表能级,ψi ( r )代表Kohn-Sham单体波函数.在KSDFT的理论框架里需要对交换关联泛函项进行近似处理,而第一性原理的计算精度往往取决于对交换关联泛函近似的好坏程度.除了早期发展的LDA泛函,目前较为常用的交换关联泛函是GGA泛函,例如Perdew-Burke-Ernzerhof (PBE)泛函[23].此外,Meta-GGA泛函(例如strongly constrained and appropriately-normed (SCAN)泛函[24])近年来也获得了较为广泛的应用[38,39].杂化泛函在一些体系性质的预测上可获得更好结果,例如半导体的能隙修正[35,36],但其计算量相比于GGA泛函也会有数量级的增加. ...
Accuracy of localized resolution of the identity in periodic hybrid functional calculations with numerical atomic orbitals
2
2020
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
... 式中,左边Hamiltonian量里的四项分别代表电子的动能算符、Hartree势、交换关联(exchange-correlation,简称xc)势和外(external,简称ext)势, r 代表空间坐标,n( r )代表电子密度,εi 代表能级,ψi ( r )代表Kohn-Sham单体波函数.在KSDFT的理论框架里需要对交换关联泛函项进行近似处理,而第一性原理的计算精度往往取决于对交换关联泛函近似的好坏程度.除了早期发展的LDA泛函,目前较为常用的交换关联泛函是GGA泛函,例如Perdew-Burke-Ernzerhof (PBE)泛函[23].此外,Meta-GGA泛函(例如strongly constrained and appropriately-normed (SCAN)泛函[24])近年来也获得了较为广泛的应用[38,39].杂化泛函在一些体系性质的预测上可获得更好结果,例如半导体的能隙修正[35,36],但其计算量相比于GGA泛函也会有数量级的增加. ...
PYATB: An efficient python package for electronic structure calculations using ab initio tight-binding model
1
2023
... ABACUS (atomic-orbital based ab-initio computation at UStc)是一款主要由C++语言编写的国产开源DFT软件,中文名为原子算筹.ABACUS支持平面波(plane wave)和数值原子轨道(numerical atomic orbitals) 2种基矢量下的Kohn-Sham方程求解,支持周期性边界条件和Brillouin区多k点采样算法,也支持晶格结构的对称性分析功能.程序采用模守恒赝势[21] (平面波基组还支持超软赝势),支持局域密度近似(local density approximation,LDA)[22]、广义梯度近似(generalized gradient approximation,GGA)[23]、Meta-GGA[24]、杂化泛函[25,26]等交换关联泛函,也支持外加电场、自旋轨道耦合、+U算法、van der Waals力修正、溶剂模型(solvation model)和极化修正(dipole correction)等功能.此外,开发团队基于平面波基组实现了无轨道密度泛函理论(orbital-free DFT)和随机波函数密度泛函理论(stochastic DFT)功能.基于数值原子轨道基组实现了含时演化的密度泛函理论.目前ABACUS的功能已较为齐全,可适用于从小体系到上千原子体系的电子结构优化、原子结构弛豫、分子动力学模拟等计算,支持DeePMD[2]、DP-GEN[27]、DeePKS[6]、DeepH[8]等多种机器学习辅助的电子结构算法并提供了相关接口,也提供了与ASE[28]、Phonopy[29]、Wannier90[30]、ShengBTE[31]、Hefei-NAMD[32]、PEXSI[33]、USPEX[34]、LibRI[35,36]、PYATB[37]等其他电子结构软件的接口. ...
Ab initio theory and modeling of water
1
2017
... 式中,左边Hamiltonian量里的四项分别代表电子的动能算符、Hartree势、交换关联(exchange-correlation,简称xc)势和外(external,简称ext)势, r 代表空间坐标,n( r )代表电子密度,εi 代表能级,ψi ( r )代表Kohn-Sham单体波函数.在KSDFT的理论框架里需要对交换关联泛函项进行近似处理,而第一性原理的计算精度往往取决于对交换关联泛函近似的好坏程度.除了早期发展的LDA泛函,目前较为常用的交换关联泛函是GGA泛函,例如Perdew-Burke-Ernzerhof (PBE)泛函[23].此外,Meta-GGA泛函(例如strongly constrained and appropriately-normed (SCAN)泛函[24])近年来也获得了较为广泛的应用[38,39].杂化泛函在一些体系性质的预测上可获得更好结果,例如半导体的能隙修正[35,36],但其计算量相比于GGA泛函也会有数量级的增加. ...
Versatile van der Waals density functional based on a meta-generalized gradient approximation
1
2016
... 式中,左边Hamiltonian量里的四项分别代表电子的动能算符、Hartree势、交换关联(exchange-correlation,简称xc)势和外(external,简称ext)势, r 代表空间坐标,n( r )代表电子密度,εi 代表能级,ψi ( r )代表Kohn-Sham单体波函数.在KSDFT的理论框架里需要对交换关联泛函项进行近似处理,而第一性原理的计算精度往往取决于对交换关联泛函近似的好坏程度.除了早期发展的LDA泛函,目前较为常用的交换关联泛函是GGA泛函,例如Perdew-Burke-Ernzerhof (PBE)泛函[23].此外,Meta-GGA泛函(例如strongly constrained and appropriately-normed (SCAN)泛函[24])近年来也获得了较为广泛的应用[38,39].杂化泛函在一些体系性质的预测上可获得更好结果,例如半导体的能隙修正[35,36],但其计算量相比于GGA泛函也会有数量级的增加. ...
Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients
1
1992
... 平面波基组加上赝势[40]是KSDFT方法中被较为广泛使用的方法.平面波基组是一组正交基,天然适合描述具有周期性边界条件的体系.采用平面波基组进行材料计算时,只需设置电子动能截断值就可定义计算所需的平面波基组,“动能”低于能量截断值的平面波会被选中,而更高频的平面波则被忽略.选取的能量截断值越高,则平面波基组越完备且精度越高,但同时计算量也会增加,因此用户在进行DFT计算前需对能量截断值做收敛性测试.此外,具有周期性边界条件的体系可定义相应的第一Brillouin区,该区域内的点称为k点.对于大体系,第一Brillouin区较小,可采用坐标为(0, 0, 0)的k点(即Gamma点)进行计算.然而,对于小体系需用更多k点.由于每个k点的Kohn-Sham方程需单独求解,因此DFT的计算量也会随k点增加而上升,在进行DFT计算前也需对k点进行收敛性测试. ...
Projector augmented-wave method
1
1994
... 模守恒赝势(norm-conserving pseudopotentials)[21]是一种广泛应用于描述电子-离子相互作用的近似方法,其目的是在保证计算精度的同时降低计算成本.模守恒赝势通过求解单原子Kohn-Sham方程,获得给定交换关联泛函近似下的原子核与价电子相互作用的近似表达,有效减少了求解Kohn-Sham方程所需电子数,尤其是对高Z (Z为原子序数)元素而言,可极大减少由于考虑全部电子而导致的较大计算量.此外,如果赝势里包含了非局域赝势,则需用到单电子Kohn-Sham波函数.模守恒赝势的构造方式有多种,不同赝势所描述的价电子数量可能不同.一般而言,包含价电子数少的赝势会使DFT计算效率更高,但降低了精度.反之,如果包含的价电子数较多,虽然保证了计算的精度但也降低了计算效率.因此,用户在选择使用赝势的时候需对精度和效率做一定的权衡.与projector augmented-wave (PAW)方法[41]相比,模守恒赝势所采用的能量截断值更高,因此PAW在计算效率方面有优势,但基于PAW的电子结构算法的实现过程相比于模守恒赝势更加复杂.目前,ABACUS软件支持多种格式的模守恒赝势,例如SG15模守恒赝势和Dojo模守恒赝势等,以及支持平面波基组下的超软赝势[42]. ...
Soft self-consistent pseudopotentials in a generalized eigenvalue formalism
1
1990
... 模守恒赝势(norm-conserving pseudopotentials)[21]是一种广泛应用于描述电子-离子相互作用的近似方法,其目的是在保证计算精度的同时降低计算成本.模守恒赝势通过求解单原子Kohn-Sham方程,获得给定交换关联泛函近似下的原子核与价电子相互作用的近似表达,有效减少了求解Kohn-Sham方程所需电子数,尤其是对高Z (Z为原子序数)元素而言,可极大减少由于考虑全部电子而导致的较大计算量.此外,如果赝势里包含了非局域赝势,则需用到单电子Kohn-Sham波函数.模守恒赝势的构造方式有多种,不同赝势所描述的价电子数量可能不同.一般而言,包含价电子数少的赝势会使DFT计算效率更高,但降低了精度.反之,如果包含的价电子数较多,虽然保证了计算的精度但也降低了计算效率.因此,用户在选择使用赝势的时候需对精度和效率做一定的权衡.与projector augmented-wave (PAW)方法[41]相比,模守恒赝势所采用的能量截断值更高,因此PAW在计算效率方面有优势,但基于PAW的电子结构算法的实现过程相比于模守恒赝势更加复杂.目前,ABACUS软件支持多种格式的模守恒赝势,例如SG15模守恒赝势和Dojo模守恒赝势等,以及支持平面波基组下的超软赝势[42]. ...
DPA-2: Towards a universal large atomic model for molecular and material simulation
2
2312
... 在平面波基组的Kohn-Sham方程求解过程中,最耗时的步骤往往来自于矩阵对角化,其计算复杂度一般为O(N3),其中N是体系原子数或电子数.受限于平面波基组下矩阵对角化的计算时间复杂度,该方法在多数场景下只适合用于处理不超过数百原子的体系.此外,平面波基组包含的基矢量数量较多,实际计算中由于内存限制难以存下整个Hamiltonian矩阵,通常使用迭代法来计算本征值和本征向量.近年来,图形处理器(GPU)硬件在科学计算中被广泛使用,ABACUS团队在GPU和曙光深度计算处理器(DCU)硬件上实现了基于平面波基组的自洽迭代算法,获得了良好的加速效果,该方法已在实际材料计算中(如DPA-2大原子模型中[43])被大量使用. ...
... 2024年,Liu等[77]采用ABACUS生成训练数据,生成了IIB-VIA族半导体材料的大原子机器学习势函数模型DPA-Semi.如图2[77]所示,该模型共计包含19种半导体材料(Si、Ge、SiC、BAs、BN、AlN、AlP、AlAs、InP、InAs、InSb、GaN、GaP、GaAs、CdTe、InTe、CdSe、ZnS和CdS).他们测试了不同半导体的晶格常数、体模量、剪切模量和Young's模量等性质,计算结果表明DPA-Semi模型的计算结果与DFT计算值一致.此外,他们利用DPA-Semi模型研究了各种半导体材料的声子谱、液体和非晶结构以及熔化温度,结果与实验和其他计算工作的结果一致,验证了DPA-Semi的精度和可靠性.将半导体体系拓展到金属体系也具有可行性,并且已经初步在DPA-2模型中实现[43]. ...
Ab initio electronic structure calculations based on numerical atomic orbitals: Basic fomalisms and recent progresses
1
2024
... 除了采用平面波作为基矢量,KSDFT方法还可采用数值原子轨道作为基矢量.数值原子轨道可以写成径向部分函数和球谐函数的乘积[44]: ...
Numerical atomic orbitals for linear-scaling calculations
1
2001
... 构造精度高、可系统提升数量、可移植性好的原子轨道基组颇具挑战.Junquera等[45]提出在一维量子力学方程中加入约束场,从而求解出具有严格截断的数值原子轨道.Ozaki[19]在OpenMX软件中采用变分法优化局域轨道形状.Blum等[20]提出在全电子DFT软件FHI-aims中,可在一个大的局域轨道基组池中挑选最优的局域轨道,从而构建不同精度的基组.Chen等[46,47]提出利用溢出函数(spillage function)构造可以系统提高轨道数量的数值原子轨道,其中每个数值原子轨道的径向部分由一组球Bessel函数展开.通过对半导体、氧化物、金属、团簇等一系列体系的测试,Li等[48]验证了利用溢出函数的方法产生数值原子轨道基组在求解Kohn-Sham方程时相比平面波基组效率更高,同时保持了良好的精度.Zheng等[49]在ABACUS中实现了基于数值原子轨道基组的高精度应力计算,测试表明相比于平面波基组,数值原子轨道基组在计算收敛应力方面更具优势.Lin等[50]提出了一种拟合数值原子轨道基组的新方案,该方案加入了对参考波函数一阶导数的拟合,进一步提升了数值原子基组的精度和效率.值得注意的是,ABACUS中的数值原子轨道需和对应的赝势一起使用,目前ABACUS官网为用户提供了赝势库和对应的数值原子轨道库文件. ...
Systematically improvable optimized atomic basis sets for ab initio calculations
1
2010
... 构造精度高、可系统提升数量、可移植性好的原子轨道基组颇具挑战.Junquera等[45]提出在一维量子力学方程中加入约束场,从而求解出具有严格截断的数值原子轨道.Ozaki[19]在OpenMX软件中采用变分法优化局域轨道形状.Blum等[20]提出在全电子DFT软件FHI-aims中,可在一个大的局域轨道基组池中挑选最优的局域轨道,从而构建不同精度的基组.Chen等[46,47]提出利用溢出函数(spillage function)构造可以系统提高轨道数量的数值原子轨道,其中每个数值原子轨道的径向部分由一组球Bessel函数展开.通过对半导体、氧化物、金属、团簇等一系列体系的测试,Li等[48]验证了利用溢出函数的方法产生数值原子轨道基组在求解Kohn-Sham方程时相比平面波基组效率更高,同时保持了良好的精度.Zheng等[49]在ABACUS中实现了基于数值原子轨道基组的高精度应力计算,测试表明相比于平面波基组,数值原子轨道基组在计算收敛应力方面更具优势.Lin等[50]提出了一种拟合数值原子轨道基组的新方案,该方案加入了对参考波函数一阶导数的拟合,进一步提升了数值原子基组的精度和效率.值得注意的是,ABACUS中的数值原子轨道需和对应的赝势一起使用,目前ABACUS官网为用户提供了赝势库和对应的数值原子轨道库文件. ...
Electronic structure interpolation via atomic orbitals
1
2011
... 构造精度高、可系统提升数量、可移植性好的原子轨道基组颇具挑战.Junquera等[45]提出在一维量子力学方程中加入约束场,从而求解出具有严格截断的数值原子轨道.Ozaki[19]在OpenMX软件中采用变分法优化局域轨道形状.Blum等[20]提出在全电子DFT软件FHI-aims中,可在一个大的局域轨道基组池中挑选最优的局域轨道,从而构建不同精度的基组.Chen等[46,47]提出利用溢出函数(spillage function)构造可以系统提高轨道数量的数值原子轨道,其中每个数值原子轨道的径向部分由一组球Bessel函数展开.通过对半导体、氧化物、金属、团簇等一系列体系的测试,Li等[48]验证了利用溢出函数的方法产生数值原子轨道基组在求解Kohn-Sham方程时相比平面波基组效率更高,同时保持了良好的精度.Zheng等[49]在ABACUS中实现了基于数值原子轨道基组的高精度应力计算,测试表明相比于平面波基组,数值原子轨道基组在计算收敛应力方面更具优势.Lin等[50]提出了一种拟合数值原子轨道基组的新方案,该方案加入了对参考波函数一阶导数的拟合,进一步提升了数值原子基组的精度和效率.值得注意的是,ABACUS中的数值原子轨道需和对应的赝势一起使用,目前ABACUS官网为用户提供了赝势库和对应的数值原子轨道库文件. ...
Large-scale ab initio simulations based on systematically improvable atomic basis
1
2016
... 构造精度高、可系统提升数量、可移植性好的原子轨道基组颇具挑战.Junquera等[45]提出在一维量子力学方程中加入约束场,从而求解出具有严格截断的数值原子轨道.Ozaki[19]在OpenMX软件中采用变分法优化局域轨道形状.Blum等[20]提出在全电子DFT软件FHI-aims中,可在一个大的局域轨道基组池中挑选最优的局域轨道,从而构建不同精度的基组.Chen等[46,47]提出利用溢出函数(spillage function)构造可以系统提高轨道数量的数值原子轨道,其中每个数值原子轨道的径向部分由一组球Bessel函数展开.通过对半导体、氧化物、金属、团簇等一系列体系的测试,Li等[48]验证了利用溢出函数的方法产生数值原子轨道基组在求解Kohn-Sham方程时相比平面波基组效率更高,同时保持了良好的精度.Zheng等[49]在ABACUS中实现了基于数值原子轨道基组的高精度应力计算,测试表明相比于平面波基组,数值原子轨道基组在计算收敛应力方面更具优势.Lin等[50]提出了一种拟合数值原子轨道基组的新方案,该方案加入了对参考波函数一阶导数的拟合,进一步提升了数值原子基组的精度和效率.值得注意的是,ABACUS中的数值原子轨道需和对应的赝势一起使用,目前ABACUS官网为用户提供了赝势库和对应的数值原子轨道库文件. ...
Accurate stress calculations based on numerical atomic orbital bases: Implementation and benchmarks
1
2021
... 构造精度高、可系统提升数量、可移植性好的原子轨道基组颇具挑战.Junquera等[45]提出在一维量子力学方程中加入约束场,从而求解出具有严格截断的数值原子轨道.Ozaki[19]在OpenMX软件中采用变分法优化局域轨道形状.Blum等[20]提出在全电子DFT软件FHI-aims中,可在一个大的局域轨道基组池中挑选最优的局域轨道,从而构建不同精度的基组.Chen等[46,47]提出利用溢出函数(spillage function)构造可以系统提高轨道数量的数值原子轨道,其中每个数值原子轨道的径向部分由一组球Bessel函数展开.通过对半导体、氧化物、金属、团簇等一系列体系的测试,Li等[48]验证了利用溢出函数的方法产生数值原子轨道基组在求解Kohn-Sham方程时相比平面波基组效率更高,同时保持了良好的精度.Zheng等[49]在ABACUS中实现了基于数值原子轨道基组的高精度应力计算,测试表明相比于平面波基组,数值原子轨道基组在计算收敛应力方面更具优势.Lin等[50]提出了一种拟合数值原子轨道基组的新方案,该方案加入了对参考波函数一阶导数的拟合,进一步提升了数值原子基组的精度和效率.值得注意的是,ABACUS中的数值原子轨道需和对应的赝势一起使用,目前ABACUS官网为用户提供了赝势库和对应的数值原子轨道库文件. ...
Strategy for constructing compact numerical atomic orbital basis sets by incorporating the gradients of reference wavefunctions
1
2021
... 构造精度高、可系统提升数量、可移植性好的原子轨道基组颇具挑战.Junquera等[45]提出在一维量子力学方程中加入约束场,从而求解出具有严格截断的数值原子轨道.Ozaki[19]在OpenMX软件中采用变分法优化局域轨道形状.Blum等[20]提出在全电子DFT软件FHI-aims中,可在一个大的局域轨道基组池中挑选最优的局域轨道,从而构建不同精度的基组.Chen等[46,47]提出利用溢出函数(spillage function)构造可以系统提高轨道数量的数值原子轨道,其中每个数值原子轨道的径向部分由一组球Bessel函数展开.通过对半导体、氧化物、金属、团簇等一系列体系的测试,Li等[48]验证了利用溢出函数的方法产生数值原子轨道基组在求解Kohn-Sham方程时相比平面波基组效率更高,同时保持了良好的精度.Zheng等[49]在ABACUS中实现了基于数值原子轨道基组的高精度应力计算,测试表明相比于平面波基组,数值原子轨道基组在计算收敛应力方面更具优势.Lin等[50]提出了一种拟合数值原子轨道基组的新方案,该方案加入了对参考波函数一阶导数的拟合,进一步提升了数值原子基组的精度和效率.值得注意的是,ABACUS中的数值原子轨道需和对应的赝势一起使用,目前ABACUS官网为用户提供了赝势库和对应的数值原子轨道库文件. ...
A caveat of the charge-extrapolation scheme for modeling electrochemical reactions on semiconductor surfaces: An issue induced by a discontinuous Fermi level change
1
2022
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
Disordered hyperuniformity in two-dimensional amorphous silica
1
2020
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
Disordered hyperuniform quasi-one-dimensional materials
1
2022
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
Peculiar band geometry induced giant shift current in ferroelectric SnTe monolayer
1
2024
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
Family behavior and Dirac bands in armchair nanoribbons with 4-8 defect lines
1
2024
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
Tuning of Berry-curvature dipole in Ta-As slabs: An effective route to enhance the nonlinear Hall response
1
2024
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
Efficient homojunction tin perovskite solar cells enabled by gradient germanium doping
1
2024
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
Interplay between magnetic structures and surface states in MnBi2Te4 from first-principles studies
1
2023
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
Retention and recycling of deuterium in liquid lithium-tin slab studied by first-principles molecular dynamics
1
2021
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
First-principles molecular dynamics study of deuterium diffusion in liquid tin
1
2017
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
Cooperative effect in a graphite intercalation compound: Enhanced mobility of AlCl4 in the graphite cathode of aluminum-ion batteries
1
2019
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
A DFT study of energetic and structural properties of a full turn of A-form DNA under relaxed and stretching conditions
1
2019
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
Copper-doped beryllium and beryllium oxide interface: A first-principles study
1
2021
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
Structure evolution of chromium-doped boron clusters: Toward the formation of endohedral boron cages
1
2019
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
Diffusion coefficients of Mg isotopes in MgSiO3 and Mg2SiO4 melts calculated by first-principles molecular dynamics simulations
1
2018
... 2022年,Liu等[51]采用ABACUS计算了大尺寸的半导体表面催化反应,将前人提出的电荷外推法应用范围从之前的金属电极表面反应拓展到半导体电极表面反应,该工作需处理最大1620个原子和11214个电子的大体系.传统基于平面波的第一性原理计算软件(例如VASP或Quantum Espresso)难以进行上千原子的大体系计算.由于ABACUS利用数值原子轨道的局域性从而构造Hamiltonian矩阵,再加上message passing interface (MPI)和OpenMP的并行化处理,因此ABACUS有能力进行千原子以上的高效计算.该工作基于SG15模守恒赝势和PBE泛函,并且采用了兼具效率和精度的double zeta plus polarization (DZP)数值原子轨道基组,作者采用共轭梯度算法进行原子结构弛豫,还使用了针对Gamma点的加速算法.通过上述参数的设置,对于1620个原子的体系(加真空),采用120个E5-2690v3的中央处理器(CPU)进程进行并行计算,单步电子迭代的时间在80 s左右.此外,ABACUS的数值原子轨道方法也在其他材料体系里被广泛使用,例如无序超均匀态的二维材料(1800个原子)[52]和准一维材料碳纳米管[53]、二维铁电材料[54]、二维石墨烯纳米带[55]、薄膜材料[56]、钙钛矿材料[57]、反铁磁拓扑绝缘体[58]、液态金属[59,60]、电池材料[61]、DNA[62]、金属界面[63]、团簇[64]、矿石中的同位素扩散[65]等. ...
Self-averaging stochastic Kohn-Sham density-functional theory
1
2013
... 2013年,Baer等[66]提出了有限温度下的随机密度泛函理论(stochastic DFT,SDFT),利用Chebysev展开以及随机轨道求迹的方法得到基态电子密度,从而省去对角化矩阵的步骤,极大地提升了效率.此后,该方法被用于结合有限温度的DFT方法,成功模拟了温稠密物质的性质并在极端高温下展现出了相较于传统有限温度KSDFT方法在效率上的绝对优势.2020年,来自美国Los Alamos国家实验室的White和Collins[67]提出了混合随机密度泛函理论(mixed stochastic-deterministic DFT,MDFT)方法,该方法在低能级采用KS轨道描述而在高能级使用随机轨道展开,因此可大大降低SDFT的随机误差.以上工作为较大尺度模拟和极端高温条件下传统KSDFT方法低效的难题提供了新的解决方法. ...
Fast and universal Kohn-Sham density functional theory algorithm for warm dense matter to hot dense plasma
1
2020
... 2013年,Baer等[66]提出了有限温度下的随机密度泛函理论(stochastic DFT,SDFT),利用Chebysev展开以及随机轨道求迹的方法得到基态电子密度,从而省去对角化矩阵的步骤,极大地提升了效率.此后,该方法被用于结合有限温度的DFT方法,成功模拟了温稠密物质的性质并在极端高温下展现出了相较于传统有限温度KSDFT方法在效率上的绝对优势.2020年,来自美国Los Alamos国家实验室的White和Collins[67]提出了混合随机密度泛函理论(mixed stochastic-deterministic DFT,MDFT)方法,该方法在低能级采用KS轨道描述而在高能级使用随机轨道展开,因此可大大降低SDFT的随机误差.以上工作为较大尺度模拟和极端高温条件下传统KSDFT方法低效的难题提供了新的解决方法. ...
Plane-wave-based stochastic-deterministic density functional theory for extended systems
2
2022
... 2022年,Liu和Chen[68]在ABACUS 2.3版本中实现了SDFT和MDFT方法,该工作采用平面波基组与模守恒赝势实现SDFT与MDFT的计算,并且首次在SDFT与MDFT算法的基础上支持Brillouin区的多k点采样,这使得周期性边界条件下的计算结果更为准确.此外,开发者针对这2种方法实现了k点和随机波函数2个层次的并行功能,并成功地应用于高效预测极端高温下的B、C和Si体系的能量、受力、压强、态密度等物理性质[68,69]. ...
... [68,69]. ...
Combining stochastic density functional theory with deep potential molecular dynamics to study warm dense matter
1
2024
... 2022年,Liu和Chen[68]在ABACUS 2.3版本中实现了SDFT和MDFT方法,该工作采用平面波基组与模守恒赝势实现SDFT与MDFT的计算,并且首次在SDFT与MDFT算法的基础上支持Brillouin区的多k点采样,这使得周期性边界条件下的计算结果更为准确.此外,开发者针对这2种方法实现了k点和随机波函数2个层次的并行功能,并成功地应用于高效预测极端高温下的B、C和Si体系的能量、受力、压强、态密度等物理性质[68,69]. ...
DeePKS: A comprehensive data-driven approach toward chemically accurate density functional theory
1
2021
... 机器学习势函数方法的瓶颈之一是在势函数的训练过程中需要生成大量的第一性原理计算数据,这些计算不仅需要较大的成本,还使得机器学习势函数的生成需要较长的周期,降低了项目快速迭代的可能性.若需使用高精度(例如杂化泛函)的第一性原理方法来产生数据,则会极大增加机器学习势函数的训练数据成本,从而限制了机器学习势函数的应用范围.针对此类问题,采用基于机器学习的DeePKS方法[70]以及相应的DeePKS-kit软件[6]可以有效解决精度和效率难两全的困境. ...
DeePKS + ABACUS as a bridge between expensive quantum mechanical models and machine learning potentials
5
2022
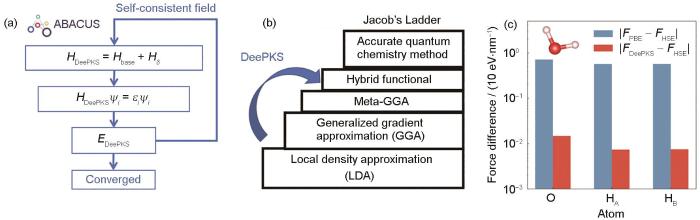
... 开发团队在ABACUS 3.0版本里推出了基于周期性边界条件的DeePKS方法[71],该算法将体系的总能量EDeePKS表示为: ...
... 式中,表示由效率较高但精度较低的基准交换关联泛函所计算得到的体系能量;而代表经过深度神经网络修正的体系能量,由体系的每一个原子贡献相加得到.图1介绍了DeePKS方法中的电子自洽迭代循环流程[71],通过深度神经网络来补充描述体系的Hamiltonian量,可以实现杂化泛函或更高精度的电子结构计算,并且以H2O分子上的原子受力为例展示了DeePKS方法的效果.图1a[71]展示了该算法通过在电子自洽迭代循环中进行LDA或GGA泛函计算得到体系Hamiltonian量Hbase,再加上已训练好的Hamiltonian量修正项Hδ 从而得到体系的总Hamiltonian量HDeePKS.进一步,对HDeePKS进行对角化计算得到电子能级{εi }和波函数{ψi },最后通过电子迭代收敛判据来计算EDeePKS.如图1b所示,从交换关联泛函的Jacob's Ladder来看,ABACUS中实现的DeePKS算法可以通过在LDA或GGA泛函的基础上引入深度神经网络的修正计算达到hybrid functional或者更准确的量子化学方法的精度.图1c展示了采用Heyd-Scuseria-Ernzerhof (HSE)交换关联泛函计算给定单个H2O分子的受力,并以此为标准计算PBE交换关联泛函和DeePKS泛函所计算的原子受力与HSE的差别,结果表明DeePKS方法的结果较好地符合HSE的结果,误差约为0.1 eV/nm. ...
... [71]展示了该算法通过在电子自洽迭代循环中进行LDA或GGA泛函计算得到体系Hamiltonian量Hbase,再加上已训练好的Hamiltonian量修正项Hδ 从而得到体系的总Hamiltonian量HDeePKS.进一步,对HDeePKS进行对角化计算得到电子能级{εi }和波函数{ψi },最后通过电子迭代收敛判据来计算EDeePKS.如图1b所示,从交换关联泛函的Jacob's Ladder来看,ABACUS中实现的DeePKS算法可以通过在LDA或GGA泛函的基础上引入深度神经网络的修正计算达到hybrid functional或者更准确的量子化学方法的精度.图1c展示了采用Heyd-Scuseria-Ernzerhof (HSE)交换关联泛函计算给定单个H2O分子的受力,并以此为标准计算PBE交换关联泛函和DeePKS泛函所计算的原子受力与HSE的差别,结果表明DeePKS方法的结果较好地符合HSE的结果,误差约为0.1 eV/nm. ...
... [
71] (The Hamiltonian value for the DeePKS and the base methods are depicted as
HDeePKS and
Hbase, respectively. In addition, the difference of the above two Hamiltonians is labeled as
Hδ . The electronic wave function and eigenvalue for the
th state are labeled as
ψi and
εi, respectively.
EDeePKS—is the total energy from the DeePKS method) (a); DeePKS algorithm can achieve the accuracy of Hybrid functionals or more precise quantum chemistry methods by incorporating corrections from deep neural networks on top of LDA or GGA functionals (b); and forces on a given single water molecule were calculated using the PBE functional (
FPBE), the Heyd-Scuseria-Ernzerhof (HSE) hybrid functional (
FHSE) and the DeePKS model (
FDeePKS), and the results showed that the DeePKS method's outcomes correspond well with those from the HSE. The label O refers to the oxygen atom while H
A and H
B refer to the two hydrogen atoms of a water molecule, respectively (c)
Fig.1![]()
ABACUS里实现的DeePKS方法旨在结合低精度DFT泛函与神经网络修正项,通过较低的计算成本,模拟高精度第一性原理方法的计算结果.DeePKS利用神经网络修正项去学习基准泛函求解Kohn-Sham方程得到的结果(低精度、低成本)与目标第一性原理方法(高精度、高成本)的结果之差.具体来说,这些结果包含计算得出的能量与原子受力和晶格应力之差,从而提升泛函精度.DeePKS中的神经网络修正项是一项局域修正,只需考虑原子周围的电子结构信息.因此,修正项带来的额外计算成本与低精度DFT泛函相当,并显著低于高精度的第一性原理方法.DeePKS拥有较强的泛化能力,训练DeePKS模型所需的数据集小于训练机器学习势函数所需数据集,有望大幅压缩数据生产成本,这将有利于推动更高精度机器学习势函数的开发,并扩展机器学习势函数的应用场景.ABACUS结合DeePKS方法已被用于研究液态水和盐水的结构和动力学性质[71],并取得了合理的结果.DeePKS方法还被用于准确预测卤化物钙钛矿的能带带隙[72],有助于设计高效稳定的太阳能电池.此外,DeePKS还被用于深入理解溶液中的质子迁移机制[73]. ...
... ABACUS里实现的DeePKS方法旨在结合低精度DFT泛函与神经网络修正项,通过较低的计算成本,模拟高精度第一性原理方法的计算结果.DeePKS利用神经网络修正项去学习基准泛函求解Kohn-Sham方程得到的结果(低精度、低成本)与目标第一性原理方法(高精度、高成本)的结果之差.具体来说,这些结果包含计算得出的能量与原子受力和晶格应力之差,从而提升泛函精度.DeePKS中的神经网络修正项是一项局域修正,只需考虑原子周围的电子结构信息.因此,修正项带来的额外计算成本与低精度DFT泛函相当,并显著低于高精度的第一性原理方法.DeePKS拥有较强的泛化能力,训练DeePKS模型所需的数据集小于训练机器学习势函数所需数据集,有望大幅压缩数据生产成本,这将有利于推动更高精度机器学习势函数的开发,并扩展机器学习势函数的应用场景.ABACUS结合DeePKS方法已被用于研究液态水和盐水的结构和动力学性质[71],并取得了合理的结果.DeePKS方法还被用于准确预测卤化物钙钛矿的能带带隙[72],有助于设计高效稳定的太阳能电池.此外,DeePKS还被用于深入理解溶液中的质子迁移机制[73]. ...
DeePKS model for halide perovskites with the accuracy of a hybrid functional
1
2023
... ABACUS里实现的DeePKS方法旨在结合低精度DFT泛函与神经网络修正项,通过较低的计算成本,模拟高精度第一性原理方法的计算结果.DeePKS利用神经网络修正项去学习基准泛函求解Kohn-Sham方程得到的结果(低精度、低成本)与目标第一性原理方法(高精度、高成本)的结果之差.具体来说,这些结果包含计算得出的能量与原子受力和晶格应力之差,从而提升泛函精度.DeePKS中的神经网络修正项是一项局域修正,只需考虑原子周围的电子结构信息.因此,修正项带来的额外计算成本与低精度DFT泛函相当,并显著低于高精度的第一性原理方法.DeePKS拥有较强的泛化能力,训练DeePKS模型所需的数据集小于训练机器学习势函数所需数据集,有望大幅压缩数据生产成本,这将有利于推动更高精度机器学习势函数的开发,并扩展机器学习势函数的应用场景.ABACUS结合DeePKS方法已被用于研究液态水和盐水的结构和动力学性质[71],并取得了合理的结果.DeePKS方法还被用于准确预测卤化物钙钛矿的能带带隙[72],有助于设计高效稳定的太阳能电池.此外,DeePKS还被用于深入理解溶液中的质子迁移机制[73]. ...
Intramolecular and water mediated tautomerism of solvated glycine
1
2024
... ABACUS里实现的DeePKS方法旨在结合低精度DFT泛函与神经网络修正项,通过较低的计算成本,模拟高精度第一性原理方法的计算结果.DeePKS利用神经网络修正项去学习基准泛函求解Kohn-Sham方程得到的结果(低精度、低成本)与目标第一性原理方法(高精度、高成本)的结果之差.具体来说,这些结果包含计算得出的能量与原子受力和晶格应力之差,从而提升泛函精度.DeePKS中的神经网络修正项是一项局域修正,只需考虑原子周围的电子结构信息.因此,修正项带来的额外计算成本与低精度DFT泛函相当,并显著低于高精度的第一性原理方法.DeePKS拥有较强的泛化能力,训练DeePKS模型所需的数据集小于训练机器学习势函数所需数据集,有望大幅压缩数据生产成本,这将有利于推动更高精度机器学习势函数的开发,并扩展机器学习势函数的应用场景.ABACUS结合DeePKS方法已被用于研究液态水和盐水的结构和动力学性质[71],并取得了合理的结果.DeePKS方法还被用于准确预测卤化物钙钛矿的能带带隙[72],有助于设计高效稳定的太阳能电池.此外,DeePKS还被用于深入理解溶液中的质子迁移机制[73]. ...
Active learning of uniformly accurate interatomic potentials for materials simulation
1
2019
... 此外ABACUS 3.0版本还支持与其他AI算法结合,例如支持主动学习生成机器学习势函数的DP-GEN[74]方法,以及支持基于原子位置生成Hamiltonian矩阵的DeepH[8]方法,这些新算法可以进一步提升电子和原子尺度的计算精度和效率. ...
Pretraining of attention-based deep learning potential model for molecular simulation
1
2024
... 式中包含了第一性原理和深度势能所预测的体系能量Δϵ、原子受力ΔFi 和Virial张量Δ的区别,其中pε、pf、p分别代表这3项的权重系数.在势函数的应用端,当用户产生了一个机器学习势函数之后,往往希望扩大应用场景进行深入计算,从而获得对材料性质更全面的认识.而在势函数的生成端,当生成的机器学习势函数模型数量增多之后,也会随之积累大量第一性原理数据,但这些数据所用的参数标准可能不完全相同,很难合在一起训练.DPA预训练大原子模型应运而生[75],其目标是使用多来源第一性原理数据训练一个可面向多元素的“预训练大模型”,从而在实际使用时只需添加少量的数据进行模型微调即可用于新材料研究. ...
Universal interatomic potential for perovskite oxides
1
2023
... 2023年,Wu等[76]采用ABACUS为适用于钙钛矿氧化物的通用力场UniPero产生训练数据.研究团队构建了一个较为精炼的训练数据集(接近20000个构型),涉及14种金属元素、26种不同类型钙钛矿氧化物的200多种组分.得益于DPA-1强大的可表示性和可迁移性,UniPero模型能较好地拟合训练数据集中的体系能量和原子受力,这些数据由ABACUS软件求解Kohn-Sham方程获得.进一步测试表明,UniPero不仅能实现对由多种元素组成的钙钛矿氧化物及其任意组分固溶体的分子动力学模拟,还能够准确预测多种铁电氧化物在升温过程中的相变顺序,包括复杂的三元弛豫铁电固溶体.此外,研究团队已公布用于钙钛矿氧化物的训练数据集、力场模型以及相应的力场性能自动测试流程,为UniPero的进一步发展与完善提供支持. ...
Machine-learning-based interatomic potentials for group IIB to VIA semiconductors: Toward a universal model
4
2024
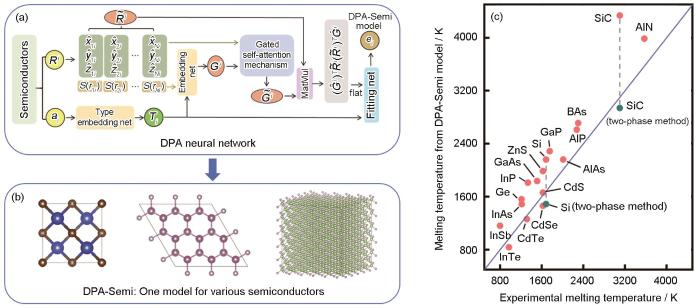
... 2024年,Liu等[77]采用ABACUS生成训练数据,生成了IIB-VIA族半导体材料的大原子机器学习势函数模型DPA-Semi.如图2[77]所示,该模型共计包含19种半导体材料(Si、Ge、SiC、BAs、BN、AlN、AlP、AlAs、InP、InAs、InSb、GaN、GaP、GaAs、CdTe、InTe、CdSe、ZnS和CdS).他们测试了不同半导体的晶格常数、体模量、剪切模量和Young's模量等性质,计算结果表明DPA-Semi模型的计算结果与DFT计算值一致.此外,他们利用DPA-Semi模型研究了各种半导体材料的声子谱、液体和非晶结构以及熔化温度,结果与实验和其他计算工作的结果一致,验证了DPA-Semi的精度和可靠性.将半导体体系拓展到金属体系也具有可行性,并且已经初步在DPA-2模型中实现[43]. ...
... [77]所示,该模型共计包含19种半导体材料(Si、Ge、SiC、BAs、BN、AlN、AlP、AlAs、InP、InAs、InSb、GaN、GaP、GaAs、CdTe、InTe、CdSe、ZnS和CdS).他们测试了不同半导体的晶格常数、体模量、剪切模量和Young's模量等性质,计算结果表明DPA-Semi模型的计算结果与DFT计算值一致.此外,他们利用DPA-Semi模型研究了各种半导体材料的声子谱、液体和非晶结构以及熔化温度,结果与实验和其他计算工作的结果一致,验证了DPA-Semi的精度和可靠性.将半导体体系拓展到金属体系也具有可行性,并且已经初步在DPA-2模型中实现[43]. ...
... [
77]
DPA, a large model based on deep neural networks, is used to train potential functions that can describe various semiconductor materials, where the first-principles training data all come from ABACUS calculations (a); DPA-Semi model generated through training also serves as an interatomic potential function, which can be used in various semiconductor simulations (b); and using the DPA-Semi model, the computed melting points of 19 semiconductors (Si, Ge, SiC, BAs, BN, AlN, AlP, AlAs, InP, InAs, InSb, GaN, GaP, GaAs, CdTe, InTe, CdSe, ZnS, CdS) obtained by the direct heating method (red dots) and the two-phase method (green dots) are relatively close to the experimental melting points (c)<sup>[<xref ref-type="bibr" rid="R77">77</xref>]</sup>Fig.2![]() <strong>2.3</strong> 基于深度学习的电子动能密度泛函
<strong>2.3</strong> 基于深度学习的电子动能密度泛函无轨道密度泛函理论(OFDFT)[78,79]是除KSDFT之外的另一种DFT算法,其中“无轨道”意味着在DFT求解基态电子密度的过程中,不采用KSDFT中的单电子Kohn-Sham轨道,因此电子动能无法由Kohn-Sham轨道计算获得,而是需要构建一个描述电子动能的动能密度泛函(kinetic energy density functional,KEDF)来计算电子动能.实际的OFDFT计算中省去了KSDFT里需要的矩阵对角化步骤,可以直接采用截断Newton法、共轭梯度法等方法优化电荷密度,从而求解能量泛函的极小值.因此OFDFT的计算复杂度远小于KSDFT,其计算体系可达百万原子级别或更大[80]. ...
... [
77]
Fig.2![]() <strong>2.3</strong> 基于深度学习的电子动能密度泛函
<strong>2.3</strong> 基于深度学习的电子动能密度泛函无轨道密度泛函理论(OFDFT)[78,79]是除KSDFT之外的另一种DFT算法,其中“无轨道”意味着在DFT求解基态电子密度的过程中,不采用KSDFT中的单电子Kohn-Sham轨道,因此电子动能无法由Kohn-Sham轨道计算获得,而是需要构建一个描述电子动能的动能密度泛函(kinetic energy density functional,KEDF)来计算电子动能.实际的OFDFT计算中省去了KSDFT里需要的矩阵对角化步骤,可以直接采用截断Newton法、共轭梯度法等方法优化电荷密度,从而求解能量泛函的极小值.因此OFDFT的计算复杂度远小于KSDFT,其计算体系可达百万原子级别或更大[80]. ...
Orbital-free kinetic-energy functionals for the nearly free electron gas
1
1998
... 无轨道密度泛函理论(OFDFT)[78,79]是除KSDFT之外的另一种DFT算法,其中“无轨道”意味着在DFT求解基态电子密度的过程中,不采用KSDFT中的单电子Kohn-Sham轨道,因此电子动能无法由Kohn-Sham轨道计算获得,而是需要构建一个描述电子动能的动能密度泛函(kinetic energy density functional,KEDF)来计算电子动能.实际的OFDFT计算中省去了KSDFT里需要的矩阵对角化步骤,可以直接采用截断Newton法、共轭梯度法等方法优化电荷密度,从而求解能量泛函的极小值.因此OFDFT的计算复杂度远小于KSDFT,其计算体系可达百万原子级别或更大[80]. ...
Kinetic-energy functional of the electron density
2
1992
... 无轨道密度泛函理论(OFDFT)[78,79]是除KSDFT之外的另一种DFT算法,其中“无轨道”意味着在DFT求解基态电子密度的过程中,不采用KSDFT中的单电子Kohn-Sham轨道,因此电子动能无法由Kohn-Sham轨道计算获得,而是需要构建一个描述电子动能的动能密度泛函(kinetic energy density functional,KEDF)来计算电子动能.实际的OFDFT计算中省去了KSDFT里需要的矩阵对角化步骤,可以直接采用截断Newton法、共轭梯度法等方法优化电荷密度,从而求解能量泛函的极小值.因此OFDFT的计算复杂度远小于KSDFT,其计算体系可达百万原子级别或更大[80]. ...
... 在凝聚态体系或分子体系中,电子动能与体系总能量在同一量级,因此OFDFT的精度依赖于电子动能泛函的精度.目前已有的电子动能泛函大多适用于简单金属(如Li、Mg、Al)及其合金,比如Wang-Teter KEDF[79]、Wang-Govind-Carter KEDF[81]等,以及简单的半导体(如Si体系),比如Huang-Carter KEDF[82].虽然OFDFT的精度低于KSDFT,但由于其效率上的优势,已被应用于合金、液态金属、团簇、温稠密物质等体系.如何设计出高精度的动能泛函仍是一个前沿基础科学问题. ...
Petascale orbital-free density functional theory enabled by small-box algorithms
1
2016
... 无轨道密度泛函理论(OFDFT)[78,79]是除KSDFT之外的另一种DFT算法,其中“无轨道”意味着在DFT求解基态电子密度的过程中,不采用KSDFT中的单电子Kohn-Sham轨道,因此电子动能无法由Kohn-Sham轨道计算获得,而是需要构建一个描述电子动能的动能密度泛函(kinetic energy density functional,KEDF)来计算电子动能.实际的OFDFT计算中省去了KSDFT里需要的矩阵对角化步骤,可以直接采用截断Newton法、共轭梯度法等方法优化电荷密度,从而求解能量泛函的极小值.因此OFDFT的计算复杂度远小于KSDFT,其计算体系可达百万原子级别或更大[80]. ...
Orbital-free kinetic-energy density functionals with a density-dependent kernel
1
1999
... 在凝聚态体系或分子体系中,电子动能与体系总能量在同一量级,因此OFDFT的精度依赖于电子动能泛函的精度.目前已有的电子动能泛函大多适用于简单金属(如Li、Mg、Al)及其合金,比如Wang-Teter KEDF[79]、Wang-Govind-Carter KEDF[81]等,以及简单的半导体(如Si体系),比如Huang-Carter KEDF[82].虽然OFDFT的精度低于KSDFT,但由于其效率上的优势,已被应用于合金、液态金属、团簇、温稠密物质等体系.如何设计出高精度的动能泛函仍是一个前沿基础科学问题. ...
Nonlocal orbital-free kinetic energy density functional for semiconductors
1
2010
... 在凝聚态体系或分子体系中,电子动能与体系总能量在同一量级,因此OFDFT的精度依赖于电子动能泛函的精度.目前已有的电子动能泛函大多适用于简单金属(如Li、Mg、Al)及其合金,比如Wang-Teter KEDF[79]、Wang-Govind-Carter KEDF[81]等,以及简单的半导体(如Si体系),比如Huang-Carter KEDF[82].虽然OFDFT的精度低于KSDFT,但由于其效率上的优势,已被应用于合金、液态金属、团簇、温稠密物质等体系.如何设计出高精度的动能泛函仍是一个前沿基础科学问题. ...
Transferable local pseudopotentials for magnesium, aluminum and silicon
1
2008
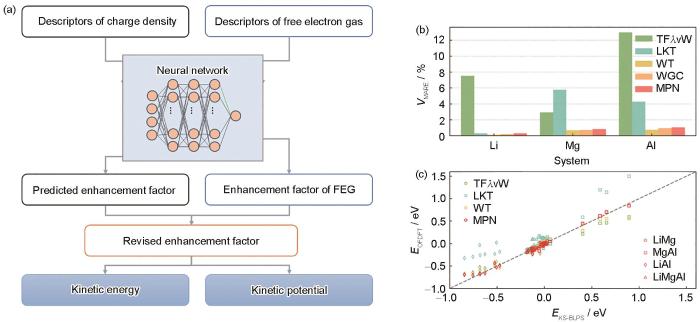
... ABACUS实现了基于平面波基组的OFDFT算法.由于缺少单电子轨道,OFDFT无法利用单电子轨道计算赝势的非局域部分,因此一般采用局域赝势.目前ABACUS支持的局域赝势为bulk-derived local pseudopotentials (BLPS)[83].如图3[84]所示,Sun等使用基于局域赝势和平面波基组,实现了一种基于机器学习且满足物理限制的非局域动能泛函(machine-learning-based physical-constrained non-local KEDF,MPN KEDF).该动能泛函满足3个物理限制:标度率、Pauli能密度的非负性及自由电子气极限,作者对Li、Mg、Al金属以及59种合金进行了测试,获得了较好的结果,为基于机器学习的电子动能密度泛函构建提供了新的思路. ...
Machine learning based nonlocal kinetic energy density functional for simple metals and alloys
3
2024
... ABACUS实现了基于平面波基组的OFDFT算法.由于缺少单电子轨道,OFDFT无法利用单电子轨道计算赝势的非局域部分,因此一般采用局域赝势.目前ABACUS支持的局域赝势为bulk-derived local pseudopotentials (BLPS)[83].如图3[84]所示,Sun等使用基于局域赝势和平面波基组,实现了一种基于机器学习且满足物理限制的非局域动能泛函(machine-learning-based physical-constrained non-local KEDF,MPN KEDF).该动能泛函满足3个物理限制:标度率、Pauli能密度的非负性及自由电子气极限,作者对Li、Mg、Al金属以及59种合金进行了测试,获得了较好的结果,为基于机器学习的电子动能密度泛函构建提供了新的思路. ...
... [
84]
Based on deep learning methods, an electronic kinetic energy density functional for machine learning is constructed, thus obtaining the electron kinetic energy and electron kinetic energy potential functions. This function introduces descriptors of free electron gas (FEG) and ensures that the electron kinetic energy satisfies physical conditions such as the free electron gas limit (a); new electronic kinetic energy functional machine-learning-based physical-constrained nonlocal (MPN), when applied to metallic systems such as Li, Mg, and Al, attained equilibrium volumes similar to those obtained with the Wang-Teter (WT) and Wang-Godvind-Carter (WGC) electronic kinetic energy functionals and demonstrated superior performance compared to other functionals like the TF<span class="formulaText"><inline-formula><math id="M22"><mi>λ</mi></math></span></inline-formula></span>vW and Luo-Karasiev-Trickey (LKT) functionals. Mean absolute relative error (MARE) of equilibrium volume is shown (<i>V</i>—equilibrium volume) (b); and MPN function has been applied to a variety of alloy systems to predict their formation energy, and compared to the results of the Kohn-Sham density functional theory, MPN exhibits better predictive performance (<i>E</i>—formation energy, OFDFT and KS-BLPS stand for orbital-free density functional theory and Kohn-Sham method with the bulk-derived local pseudopotentials, respectively) (c)<sup>[<xref ref-type="bibr" rid="R84">84</xref>]</sup>Fig.3![]() <strong>3</strong> 软件开发<strong>3.1</strong> 密度泛函理论软件的特点
<strong>3</strong> 软件开发<strong>3.1</strong> 密度泛函理论软件的特点密度泛函理论软件的开发具有较大的复杂度.首先是软件的功能庞杂,输入参数多.以支持平面波基组和赝势的软件为例,若软件支持自旋非极化和自旋极化2种功能,其中每一种功能下支持LDA、GGA、Meta-GGA和hybrid functional 4种交换关联泛函,并且可以让用户选择使用模守恒赝势或超软赝势,则每次开发者对软件做出修改后,都需要让更新后的软件通过2 × 4 × 2 = 16个功能测试. ...
... [
84]
Fig.3![]() <strong>3</strong> 软件开发<strong>3.1</strong> 密度泛函理论软件的特点
<strong>3</strong> 软件开发<strong>3.1</strong> 密度泛函理论软件的特点密度泛函理论软件的开发具有较大的复杂度.首先是软件的功能庞杂,输入参数多.以支持平面波基组和赝势的软件为例,若软件支持自旋非极化和自旋极化2种功能,其中每一种功能下支持LDA、GGA、Meta-GGA和hybrid functional 4种交换关联泛函,并且可以让用户选择使用模守恒赝势或超软赝势,则每次开发者对软件做出修改后,都需要让更新后的软件通过2 × 4 × 2 = 16个功能测试. ...





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}