Corresponding authors:WANG William Yi, professor, Tel:(029)88460294, E-mail:wywang@nwpu.edu.cn;LIU Zi-Kui, professor, Tel:(814)8651934, E-mail:zxl15@psu.edu
Received:2024-05-08Revised:2024-07-05
Fund supported:
Natural Science Foundation of Jiangsu Province(BK20230673) Doctor of Entrepreneurship and Innovation of Jiangsu Province(JSSCBS20221270)
Entropy is an important concept in science and is ubiquitous from quantum to astronomy. By integrating statistical mechanics, quantum mechanics, and thermodynamics, Professor Zi-Kui Liu proposed the zentropy theory, which stacks entropy over configurations. The zentropy theory takes the configurations in Gibbs' statistical mechanics of a given ensemble as the material gene with the ground state as the basic configuration and additional configurations ergodically derived from its internal degrees of freedom. In the zentropy theory, the total entropy of a system is defined as the weighted average of the entropy of each configuration plus the statistical entropy among all configurations. In this paper, the basic equations and principles of the zentropy theory are introduced, and their typical applications, including magnetic and ferroelectric transformations, thermal expansion mechanisms, and critical phenomenon prediction are outlined. Furthermore, a perspective on the development of this theory, software ecosystems, high-throughput computing, and integration with artificial intelligence is provided in this study.
针对上述的问题,刘梓葵教授团队[30]经过在材料热力学与材料动力学领域长期的思考与研究,于近期受邀在Journal of Phase Equilibria and Diffusion上发表了题为Zentropy theory for positive and negative thermal expansion的文章,在文中正式提出了一种通用的计算材料体系多尺度熵的理论框架:叠熵理论(zentropy = z + entropy,其中字母z源于Max Planck为配分函数创造的德语词Zusstandssumme,意为状态总和,也通常用于表示配分函数,该名字是刘梓葵教授在杜克大学讲座时由Josiah Roberts建议的。Zentropy即意为不同尺度/状态熵的叠加之和,因此中文译名为叠熵)。该理论一经发表,得到了Phys.org、ScienceDaily、SciTechDaily、AZOMaterials、量子认知百家号等著名科技网站的报道,量子认知百家号给出“综熵理论将是华人科学家们在传统的基础物理理论,特别是热力学基础科学领域作出的重要贡献” (该文中,作者将zentropy译为“综熵”)的高度评价。
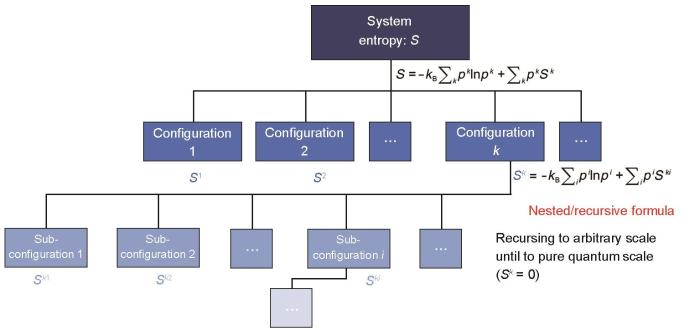
Fig.2
Schematic of zentropy theory (kB—Boltzmann constant; pk and pi —probabilities of configuration k and its sub-configuration i; Sk and Ski —entropies of configuration k and its sub-configuration i)
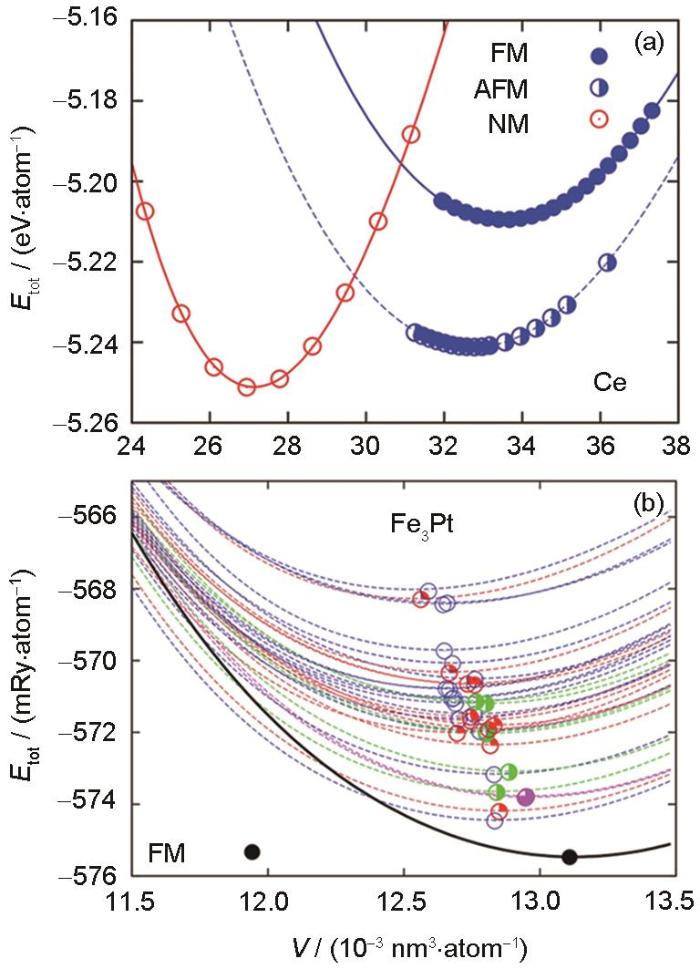
Fig.3
Volume-energy curves at 0 K of different configurations[30] (FM, AFM, and NM mean ferromagnetic, antiferromagnetic, and nonmagnetic, respectively; V and Etot mean atomic volume and total energy at 0 K, respectively)
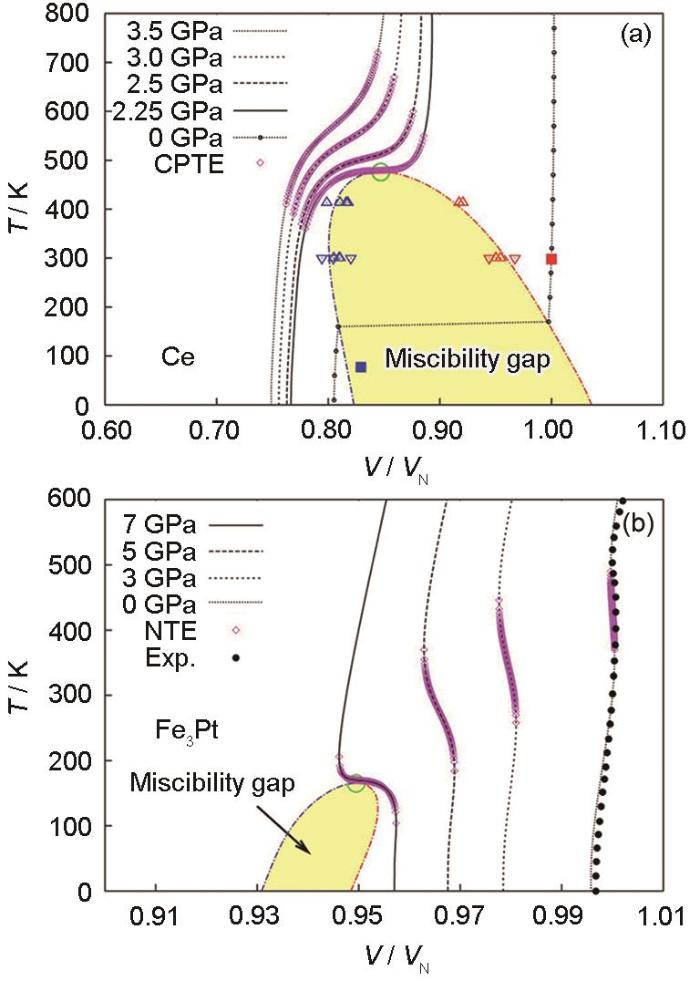
Fig.4
Temperature-volume phase diagram with isobaric volumes at various pressures[45] (T means temperature; the volume (V) is normalized to their respective equilibrium volume (VN) at atmospheric pressure and room temperature; CPTE and NTE mean colossal positive thermal expansion and negative thermal expansion, respectively; Exp. means experimental results)
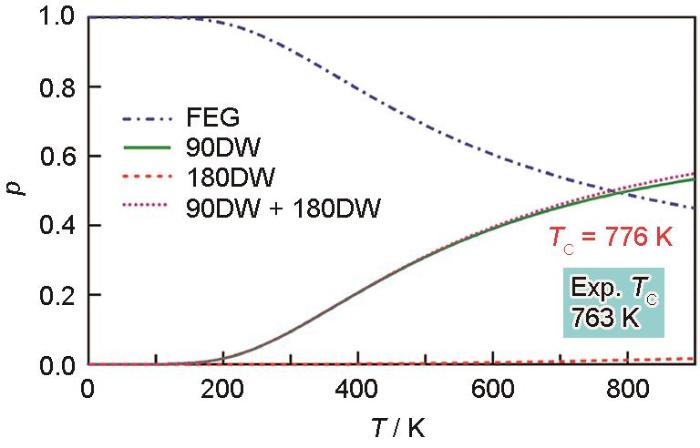
结合表1中的公式,可计算得到各个组态的概率分布,如图5[47]所示。从图中可以看出,随着温度的升高,铁电基态的概率越来越小,而非基态(尤其是90DW)的概率越来越大,当非基态组态占多数时,即铁电态占比小于50%,可作为铁电-顺电转变的判据,由此得到PbTiO3的铁电-顺电转变温度为776 K (实验结果763 K),实现了完全通过第一性原理无任何经验或者拟合参数的铁电-顺电转变临界温度的预测。此外,考虑到不同微观组态均以一定概率表现,随着温度的升高,PbTiO3中90DW的概率逐渐升高,若铁电态PbTiO3沿晶体c轴极化,则随着温度的增大,将出现大量朝向晶体a轴和b轴方向的极化(与原铁电极化方向形成90DW),在宏观上表现为PbTiO3发生从四方相到立方相的转变。通过该理论能很好地解释X射线衍射(XRD)与X射线吸收精细结构(XAFS)方法(2种方法在时间上的分辨率不同,XRD可以认为是一段时间平均的结果,而XAFS可认为是瞬时的结果)测定PbTiO3宏观和微观晶格参数的矛盾[59]。
Fig.5
Probability of configurations as a function of temperature in PbTiO3[47] (p—probability, DW—domain wall, FEG—ground state of ferroelectric without DW, TC—critical temperature, the domain wall energies for 90DW and 180DW are 35 and 132 mJ/m2, respectively, which is taken from Ref.[58])
Fig.6
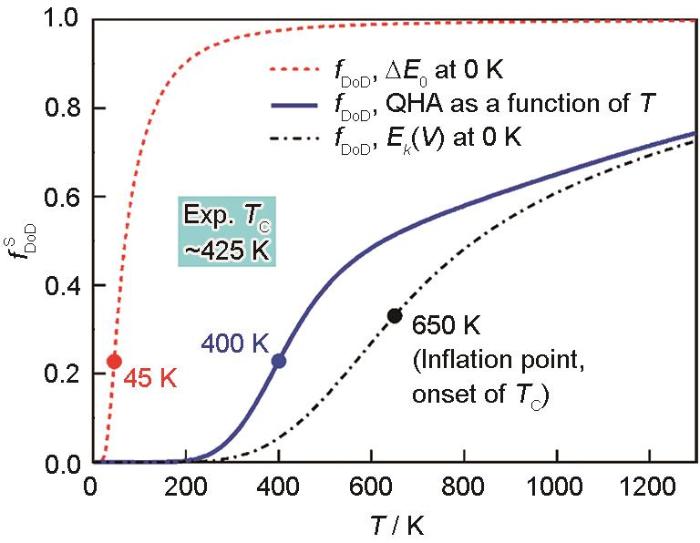
Predicted degree of disorder () as a function of temperature in Fe3Pt via entropy[46] (ΔE0 and Ek (V) are equilibrium energy and static total energy at 0 K, respectively. QHA means quasiharmonic approach, in which the free energy with contributions from vibrations and thermal electrons is evaluated)
Materials genome engineering (MGE) is a frontier technology in the field of material science and engineering, which is well capable to revolutionize the research and development (R&D) mode of new materials, greatly improve the R&D efficiency, shorten the R&D time, and reduce the cost. This paper reviews the progress of MGE in China from the aspects of the fundamental theory and methods, key technology and equipment, the R&D of new materials and related engineering application, talents training, formation and promotion of new concept of material genetic engineering. The paper also looks forward to the future development of MGE in China.
National Research Council. Integrated Computational Materials Engineering: A Transformational Discipline for Improved Competitiveness and National Security [M]. Washington, D.C.: The National Academies Press, 2008: 9
National Academies of Sciences, Engineering, and Medicine. NSF Efforts to Achieve the Nation's Vision for the Materials Genome Initiative: Designing Materials to Revolutionize and Engineer Our Future (DMREF) [M]. Washington, D.C.: The National Academies Press, 2023: 57
Metallic glasses (MGs) have attracted extensive attention in the past decades due to their unique chem-ical, physical and mechanical properties promising for a wide range of engineering applications. A thor-ough understanding of their structure-property relationships is the key to the development of novel MGs with desirable performance. New strategies, as proposed by Materials Genome Initiative (MGI), construct a new paradigm for high-throughput materials discovery and design, and are being increas-ingly implemented in the search of new MGs. While a few reports have summarized the application of high-throughput and/or machine learning techniques, a comprehensive assessment of materials genome strategies for developing MGs is still missing. Herein, this paper aims to present a timely overview of key advances in this fascinating subject, as well as current challenges and future opportunities. A holistic approach is used to cover the related topics, including high-throughput preparation and characterization of MGs, and data-driven machine learning strategies for accelerating the development of novel MGs. Fi-nally, future research directions and perspectives for MGI-assisted design of MGs are also proposed and surmised.
WangG J, PengL Y, LiK Q, et al.
ALKEMIE: An intelligent computational platform for accelerating materials discovery and design
The development of novel materials has experienced three paradigms: purely empirical, theoretical models, and computational materials science. Currently, the huge amount of data generated by experiments and simulations has facilitated a shift in materials science to a data-driven fourth paradigm. Therefore, the development of high-throughput automatic integrated computations and data mining algorithms based on material databases and artificial intelligence algorithms is critical for accelerating the design of novel materials. This paper presents an open-source distributed computational platform called Artificial Learning and Knowledge Enhanced Materials Informatics Engineering 2.0 (ALKEMIE2.0) based on the AMDIV (automation-modular-database-intelligence-visualization) design concepts. The ALKEMIE2.0 platform includes five core components of automation, modular, materials database, artificial intelligence, and visualization, which are suitable for the computational design of novel materials. The overall characteristics of ALKEMIE2.0 are divided into five pillars. ALKEMIE-Core integrates multiscale calculations and simulation software using the ALKEMIE-Plugin application programming interface. Its high-throughput calculation workflows that support 104 magnitude concurrencies are implemented by integrating the automatic frameworks of model constructions, calculation workflows, and data analyses. Furthermore, the platform is based on the ALKEMIE-Server, which can easily and automatically open daemon services and realize information interactions in distributed supercomputers. With its strong portability and scalability, ALKEMIE has been deployed in the National Supercomputing Tianjin Center. In addition, the multitype materials database called the ALKEMIE-Data Vault contains structure, task, workflow, and material property databases, which combined with the power of supercomputing, enables the rapid application of artificial intelligence algorithms in the design of new materials. In particular, the many user-friendly interfaces, which were elaborately designed using the ALKEMIE-GUI and are suitable for scientists with broad backgrounds, make structural building, work flowcharts, data analysis, and machine learning models more transparent and maneuverable. Finally, the main features of ALKEMIE2.0 are demonstrated using two examples of multiplatform deployment and high-throughput screening of binary aluminum alloys.
Discovering new materials with excellent performance is a hot issue in the materials genome initiative. Traditional experiments and calculations often waste large amounts of time and money and are also limited by various conditions. Therefore, it is imperative to develop a new method to accelerate the discovery and design of new materials. In recent years, material discovery and design methods using machine learning have attracted much attention from material experts and have made some progress. This review first outlines available materials database and material data analytics tools and then elaborates on the machine learning algorithms used in materials science. Next, the field of application of machine learning in materials science is summarized, focusing on the aspects of structure determination, performance prediction, fingerprint prediction, and new material discovery. Finally, the review points out the problems of data and machine learning in materials science and points to future research. Using machine learning algorithms, the authors hope to achieve amazing results in material discovery and design.
EW N.
AI helps to establish a new paradigm for scientific research
Density functional thermodynamic description of spin, phonon and displacement degrees of freedom in antiferromagnetic-to-paramagnetic phase transition in YNiO3
The software package ESPEI has been developed for efficient evaluation of thermodynamic model parameters within the CALPHAD method. ESPEI uses a linear fitting strategy to parameterize Gibbs energy functions of single phases based on their thermochemical data and refines the model parameters using phase equilibrium data through Bayesian parameter estimation within a Markov Chain Monte Carlo machine learning approach. In this paper, the methodologies employed in ESPEI are discussed in detail and demonstrated for the Cu-Mg system down to 0 K using unary descriptions based on segmented regression. The model parameter uncertainties are quantified and propagated to the Gibbs energy functions.
OtisR, LiuZ K.
pycalphad: CALPHAD-based computational thermodynamics in python
Phase fractions, compositions and energies of the stable phases as a function of macroscopic composition, temperature, and pressure (X-T-P) are the principle correlations needed for the design of new materials and improvement of existing materials. They are the outcomes of thermodynamic modeling based on the CALculation of PHAse Diagrams (CALPHAD) approach. The accuracy of CALPHAD predictions vary widely in X-T-P space due to experimental error, model inadequacy and unequal data coverage. In response, researchers have developed frameworks to quantify the uncertainty of thermodynamic property model parameters and propagate it to phase diagram predictions. In most previous studies, uncertainty was represented as intervals on phase boundaries (with respect to composition or temperature) and was unable to represent the uncertainty in invariant reactions or in the stability of phase regions. In this work, we propose a suite of tools that leverages samples from the multivariate model parameter distribution to represent uncertainty in forms that surpass previous limitations and are well suited to materials design. These representations include the distribution of phase diagrams and their features, as well as the dependence of phase stability and the distributions of phase fraction, composition, activity and Gibbs energy on X-T-P location - irrespective of the total number of components. Most critically, the new methodology allows the material designer to interrogate a certain composition and temperature domain and get in return the probability of different phases to be stable, which can positively impact materials design. (C) 2019 Acta Materialia Inc. Published by Elsevier Ltd.
PengJ, LeeS, WilliamsA, et al.
Advanced data science toolkit for non-data scientists—A user guide
Prospects of materials genome engineering frontiers
2
2023
... 美国提出MGI后,中国随后也建立了相应的计划:材料基因工程(materials genome engineering,MGE)[6,12,13].中国在2011年底就对MGI进行了讨论,2012年启动了“材料基因组计划”重大咨询项目,2015年编制了《材料基因工程关键技术和支撑平台重点专项实施方案》,全面开启了材料基因工程研究.在该专项的支持下,我国材料基因工程在以下方面取得了较大的进展:(1) 新材料开发与优化(如:具有优异机械性能的高温块体金属玻璃[14,15]);(2) 关键技术与装备研发(如:高通量工作流计算软件ALKEMIE[16,17],材料基因工程专用数据库平台MGEData);(3) 工程化应用示范(如:建立了重型燃气轮机专用材料数据库,推动了重型燃气轮机的数字化、智能化设计和制造[12]);(4) 创新平台建设(如:北京材料基因工程高精尖创新中心、上海大学材料基因组工程研究院、西北工业大学材料信息学与基因工程研究中心、哈尔滨工业大学(深圳)材料基因工程及大数据研究院、北京航空航天大学集成计算材料工程中心、云南稀贵金属材料基因工程创新平台[18]);(5) 国内外学术交流(如:材料基因工程高层论坛,创建了专门的刊物:Journal of Materials Informatics和MGE Advances).此外,材料基因工程大大的提高了计算方法和工具与数据库技术在材料学中的地位,由此催生了材料设计的第四范式:数据驱动的材料设计范式[19,20]和科学研究第五范式:人工智能驱动的科学(AI for Science,AI4S)[21]或智能化科研(AI for Research,AI4R)[22]. ...
... [12]);(4) 创新平台建设(如:北京材料基因工程高精尖创新中心、上海大学材料基因组工程研究院、西北工业大学材料信息学与基因工程研究中心、哈尔滨工业大学(深圳)材料基因工程及大数据研究院、北京航空航天大学集成计算材料工程中心、云南稀贵金属材料基因工程创新平台[18]);(5) 国内外学术交流(如:材料基因工程高层论坛,创建了专门的刊物:Journal of Materials Informatics和MGE Advances).此外,材料基因工程大大的提高了计算方法和工具与数据库技术在材料学中的地位,由此催生了材料设计的第四范式:数据驱动的材料设计范式[19,20]和科学研究第五范式:人工智能驱动的科学(AI for Science,AI4S)[21]或智能化科研(AI for Research,AI4R)[22]. ...
A vision of materials genome engineering in China
1
2022
... 美国提出MGI后,中国随后也建立了相应的计划:材料基因工程(materials genome engineering,MGE)[6,12,13].中国在2011年底就对MGI进行了讨论,2012年启动了“材料基因组计划”重大咨询项目,2015年编制了《材料基因工程关键技术和支撑平台重点专项实施方案》,全面开启了材料基因工程研究.在该专项的支持下,我国材料基因工程在以下方面取得了较大的进展:(1) 新材料开发与优化(如:具有优异机械性能的高温块体金属玻璃[14,15]);(2) 关键技术与装备研发(如:高通量工作流计算软件ALKEMIE[16,17],材料基因工程专用数据库平台MGEData);(3) 工程化应用示范(如:建立了重型燃气轮机专用材料数据库,推动了重型燃气轮机的数字化、智能化设计和制造[12]);(4) 创新平台建设(如:北京材料基因工程高精尖创新中心、上海大学材料基因组工程研究院、西北工业大学材料信息学与基因工程研究中心、哈尔滨工业大学(深圳)材料基因工程及大数据研究院、北京航空航天大学集成计算材料工程中心、云南稀贵金属材料基因工程创新平台[18]);(5) 国内外学术交流(如:材料基因工程高层论坛,创建了专门的刊物:Journal of Materials Informatics和MGE Advances).此外,材料基因工程大大的提高了计算方法和工具与数据库技术在材料学中的地位,由此催生了材料设计的第四范式:数据驱动的材料设计范式[19,20]和科学研究第五范式:人工智能驱动的科学(AI for Science,AI4S)[21]或智能化科研(AI for Research,AI4R)[22]. ...
High-temperature bulk metallic glasses developed by combinatorial methods
1
2019
... 美国提出MGI后,中国随后也建立了相应的计划:材料基因工程(materials genome engineering,MGE)[6,12,13].中国在2011年底就对MGI进行了讨论,2012年启动了“材料基因组计划”重大咨询项目,2015年编制了《材料基因工程关键技术和支撑平台重点专项实施方案》,全面开启了材料基因工程研究.在该专项的支持下,我国材料基因工程在以下方面取得了较大的进展:(1) 新材料开发与优化(如:具有优异机械性能的高温块体金属玻璃[14,15]);(2) 关键技术与装备研发(如:高通量工作流计算软件ALKEMIE[16,17],材料基因工程专用数据库平台MGEData);(3) 工程化应用示范(如:建立了重型燃气轮机专用材料数据库,推动了重型燃气轮机的数字化、智能化设计和制造[12]);(4) 创新平台建设(如:北京材料基因工程高精尖创新中心、上海大学材料基因组工程研究院、西北工业大学材料信息学与基因工程研究中心、哈尔滨工业大学(深圳)材料基因工程及大数据研究院、北京航空航天大学集成计算材料工程中心、云南稀贵金属材料基因工程创新平台[18]);(5) 国内外学术交流(如:材料基因工程高层论坛,创建了专门的刊物:Journal of Materials Informatics和MGE Advances).此外,材料基因工程大大的提高了计算方法和工具与数据库技术在材料学中的地位,由此催生了材料设计的第四范式:数据驱动的材料设计范式[19,20]和科学研究第五范式:人工智能驱动的科学(AI for Science,AI4S)[21]或智能化科研(AI for Research,AI4R)[22]. ...
Materials genome strategy for metallic glasses
1
2023
... 美国提出MGI后,中国随后也建立了相应的计划:材料基因工程(materials genome engineering,MGE)[6,12,13].中国在2011年底就对MGI进行了讨论,2012年启动了“材料基因组计划”重大咨询项目,2015年编制了《材料基因工程关键技术和支撑平台重点专项实施方案》,全面开启了材料基因工程研究.在该专项的支持下,我国材料基因工程在以下方面取得了较大的进展:(1) 新材料开发与优化(如:具有优异机械性能的高温块体金属玻璃[14,15]);(2) 关键技术与装备研发(如:高通量工作流计算软件ALKEMIE[16,17],材料基因工程专用数据库平台MGEData);(3) 工程化应用示范(如:建立了重型燃气轮机专用材料数据库,推动了重型燃气轮机的数字化、智能化设计和制造[12]);(4) 创新平台建设(如:北京材料基因工程高精尖创新中心、上海大学材料基因组工程研究院、西北工业大学材料信息学与基因工程研究中心、哈尔滨工业大学(深圳)材料基因工程及大数据研究院、北京航空航天大学集成计算材料工程中心、云南稀贵金属材料基因工程创新平台[18]);(5) 国内外学术交流(如:材料基因工程高层论坛,创建了专门的刊物:Journal of Materials Informatics和MGE Advances).此外,材料基因工程大大的提高了计算方法和工具与数据库技术在材料学中的地位,由此催生了材料设计的第四范式:数据驱动的材料设计范式[19,20]和科学研究第五范式:人工智能驱动的科学(AI for Science,AI4S)[21]或智能化科研(AI for Research,AI4R)[22]. ...
ALKEMIE: An intelligent computational platform for accelerating materials discovery and design
1
2021
... 美国提出MGI后,中国随后也建立了相应的计划:材料基因工程(materials genome engineering,MGE)[6,12,13].中国在2011年底就对MGI进行了讨论,2012年启动了“材料基因组计划”重大咨询项目,2015年编制了《材料基因工程关键技术和支撑平台重点专项实施方案》,全面开启了材料基因工程研究.在该专项的支持下,我国材料基因工程在以下方面取得了较大的进展:(1) 新材料开发与优化(如:具有优异机械性能的高温块体金属玻璃[14,15]);(2) 关键技术与装备研发(如:高通量工作流计算软件ALKEMIE[16,17],材料基因工程专用数据库平台MGEData);(3) 工程化应用示范(如:建立了重型燃气轮机专用材料数据库,推动了重型燃气轮机的数字化、智能化设计和制造[12]);(4) 创新平台建设(如:北京材料基因工程高精尖创新中心、上海大学材料基因组工程研究院、西北工业大学材料信息学与基因工程研究中心、哈尔滨工业大学(深圳)材料基因工程及大数据研究院、北京航空航天大学集成计算材料工程中心、云南稀贵金属材料基因工程创新平台[18]);(5) 国内外学术交流(如:材料基因工程高层论坛,创建了专门的刊物:Journal of Materials Informatics和MGE Advances).此外,材料基因工程大大的提高了计算方法和工具与数据库技术在材料学中的地位,由此催生了材料设计的第四范式:数据驱动的材料设计范式[19,20]和科学研究第五范式:人工智能驱动的科学(AI for Science,AI4S)[21]或智能化科研(AI for Research,AI4R)[22]. ...
High-throughput automatic integrated material calculations and data management intelligent platform and the application in novel alloys
1
2022
... 美国提出MGI后,中国随后也建立了相应的计划:材料基因工程(materials genome engineering,MGE)[6,12,13].中国在2011年底就对MGI进行了讨论,2012年启动了“材料基因组计划”重大咨询项目,2015年编制了《材料基因工程关键技术和支撑平台重点专项实施方案》,全面开启了材料基因工程研究.在该专项的支持下,我国材料基因工程在以下方面取得了较大的进展:(1) 新材料开发与优化(如:具有优异机械性能的高温块体金属玻璃[14,15]);(2) 关键技术与装备研发(如:高通量工作流计算软件ALKEMIE[16,17],材料基因工程专用数据库平台MGEData);(3) 工程化应用示范(如:建立了重型燃气轮机专用材料数据库,推动了重型燃气轮机的数字化、智能化设计和制造[12]);(4) 创新平台建设(如:北京材料基因工程高精尖创新中心、上海大学材料基因组工程研究院、西北工业大学材料信息学与基因工程研究中心、哈尔滨工业大学(深圳)材料基因工程及大数据研究院、北京航空航天大学集成计算材料工程中心、云南稀贵金属材料基因工程创新平台[18]);(5) 国内外学术交流(如:材料基因工程高层论坛,创建了专门的刊物:Journal of Materials Informatics和MGE Advances).此外,材料基因工程大大的提高了计算方法和工具与数据库技术在材料学中的地位,由此催生了材料设计的第四范式:数据驱动的材料设计范式[19,20]和科学研究第五范式:人工智能驱动的科学(AI for Science,AI4S)[21]或智能化科研(AI for Research,AI4R)[22]. ...
高通量自动流程集成计算与数据管理智能平台及其在合金设计中的应用
1
2022
... 美国提出MGI后,中国随后也建立了相应的计划:材料基因工程(materials genome engineering,MGE)[6,12,13].中国在2011年底就对MGI进行了讨论,2012年启动了“材料基因组计划”重大咨询项目,2015年编制了《材料基因工程关键技术和支撑平台重点专项实施方案》,全面开启了材料基因工程研究.在该专项的支持下,我国材料基因工程在以下方面取得了较大的进展:(1) 新材料开发与优化(如:具有优异机械性能的高温块体金属玻璃[14,15]);(2) 关键技术与装备研发(如:高通量工作流计算软件ALKEMIE[16,17],材料基因工程专用数据库平台MGEData);(3) 工程化应用示范(如:建立了重型燃气轮机专用材料数据库,推动了重型燃气轮机的数字化、智能化设计和制造[12]);(4) 创新平台建设(如:北京材料基因工程高精尖创新中心、上海大学材料基因组工程研究院、西北工业大学材料信息学与基因工程研究中心、哈尔滨工业大学(深圳)材料基因工程及大数据研究院、北京航空航天大学集成计算材料工程中心、云南稀贵金属材料基因工程创新平台[18]);(5) 国内外学术交流(如:材料基因工程高层论坛,创建了专门的刊物:Journal of Materials Informatics和MGE Advances).此外,材料基因工程大大的提高了计算方法和工具与数据库技术在材料学中的地位,由此催生了材料设计的第四范式:数据驱动的材料设计范式[19,20]和科学研究第五范式:人工智能驱动的科学(AI for Science,AI4S)[21]或智能化科研(AI for Research,AI4R)[22]. ...
Understanding oxidation resistance of Pt-based alloys through computations of Ellingham diagrams with experimental verifications
1
2023
... 美国提出MGI后,中国随后也建立了相应的计划:材料基因工程(materials genome engineering,MGE)[6,12,13].中国在2011年底就对MGI进行了讨论,2012年启动了“材料基因组计划”重大咨询项目,2015年编制了《材料基因工程关键技术和支撑平台重点专项实施方案》,全面开启了材料基因工程研究.在该专项的支持下,我国材料基因工程在以下方面取得了较大的进展:(1) 新材料开发与优化(如:具有优异机械性能的高温块体金属玻璃[14,15]);(2) 关键技术与装备研发(如:高通量工作流计算软件ALKEMIE[16,17],材料基因工程专用数据库平台MGEData);(3) 工程化应用示范(如:建立了重型燃气轮机专用材料数据库,推动了重型燃气轮机的数字化、智能化设计和制造[12]);(4) 创新平台建设(如:北京材料基因工程高精尖创新中心、上海大学材料基因组工程研究院、西北工业大学材料信息学与基因工程研究中心、哈尔滨工业大学(深圳)材料基因工程及大数据研究院、北京航空航天大学集成计算材料工程中心、云南稀贵金属材料基因工程创新平台[18]);(5) 国内外学术交流(如:材料基因工程高层论坛,创建了专门的刊物:Journal of Materials Informatics和MGE Advances).此外,材料基因工程大大的提高了计算方法和工具与数据库技术在材料学中的地位,由此催生了材料设计的第四范式:数据驱动的材料设计范式[19,20]和科学研究第五范式:人工智能驱动的科学(AI for Science,AI4S)[21]或智能化科研(AI for Research,AI4R)[22]. ...
Perspective: Materials informatics and big data: Realization of the “fourth paradigm” of science in materials science
1
2016
... 美国提出MGI后,中国随后也建立了相应的计划:材料基因工程(materials genome engineering,MGE)[6,12,13].中国在2011年底就对MGI进行了讨论,2012年启动了“材料基因组计划”重大咨询项目,2015年编制了《材料基因工程关键技术和支撑平台重点专项实施方案》,全面开启了材料基因工程研究.在该专项的支持下,我国材料基因工程在以下方面取得了较大的进展:(1) 新材料开发与优化(如:具有优异机械性能的高温块体金属玻璃[14,15]);(2) 关键技术与装备研发(如:高通量工作流计算软件ALKEMIE[16,17],材料基因工程专用数据库平台MGEData);(3) 工程化应用示范(如:建立了重型燃气轮机专用材料数据库,推动了重型燃气轮机的数字化、智能化设计和制造[12]);(4) 创新平台建设(如:北京材料基因工程高精尖创新中心、上海大学材料基因组工程研究院、西北工业大学材料信息学与基因工程研究中心、哈尔滨工业大学(深圳)材料基因工程及大数据研究院、北京航空航天大学集成计算材料工程中心、云南稀贵金属材料基因工程创新平台[18]);(5) 国内外学术交流(如:材料基因工程高层论坛,创建了专门的刊物:Journal of Materials Informatics和MGE Advances).此外,材料基因工程大大的提高了计算方法和工具与数据库技术在材料学中的地位,由此催生了材料设计的第四范式:数据驱动的材料设计范式[19,20]和科学研究第五范式:人工智能驱动的科学(AI for Science,AI4S)[21]或智能化科研(AI for Research,AI4R)[22]. ...
Machine learning in materials genome initiative: A review
1
2020
... 美国提出MGI后,中国随后也建立了相应的计划:材料基因工程(materials genome engineering,MGE)[6,12,13].中国在2011年底就对MGI进行了讨论,2012年启动了“材料基因组计划”重大咨询项目,2015年编制了《材料基因工程关键技术和支撑平台重点专项实施方案》,全面开启了材料基因工程研究.在该专项的支持下,我国材料基因工程在以下方面取得了较大的进展:(1) 新材料开发与优化(如:具有优异机械性能的高温块体金属玻璃[14,15]);(2) 关键技术与装备研发(如:高通量工作流计算软件ALKEMIE[16,17],材料基因工程专用数据库平台MGEData);(3) 工程化应用示范(如:建立了重型燃气轮机专用材料数据库,推动了重型燃气轮机的数字化、智能化设计和制造[12]);(4) 创新平台建设(如:北京材料基因工程高精尖创新中心、上海大学材料基因组工程研究院、西北工业大学材料信息学与基因工程研究中心、哈尔滨工业大学(深圳)材料基因工程及大数据研究院、北京航空航天大学集成计算材料工程中心、云南稀贵金属材料基因工程创新平台[18]);(5) 国内外学术交流(如:材料基因工程高层论坛,创建了专门的刊物:Journal of Materials Informatics和MGE Advances).此外,材料基因工程大大的提高了计算方法和工具与数据库技术在材料学中的地位,由此催生了材料设计的第四范式:数据驱动的材料设计范式[19,20]和科学研究第五范式:人工智能驱动的科学(AI for Science,AI4S)[21]或智能化科研(AI for Research,AI4R)[22]. ...
AI helps to establish a new paradigm for scientific research
2
2024
... 美国提出MGI后,中国随后也建立了相应的计划:材料基因工程(materials genome engineering,MGE)[6,12,13].中国在2011年底就对MGI进行了讨论,2012年启动了“材料基因组计划”重大咨询项目,2015年编制了《材料基因工程关键技术和支撑平台重点专项实施方案》,全面开启了材料基因工程研究.在该专项的支持下,我国材料基因工程在以下方面取得了较大的进展:(1) 新材料开发与优化(如:具有优异机械性能的高温块体金属玻璃[14,15]);(2) 关键技术与装备研发(如:高通量工作流计算软件ALKEMIE[16,17],材料基因工程专用数据库平台MGEData);(3) 工程化应用示范(如:建立了重型燃气轮机专用材料数据库,推动了重型燃气轮机的数字化、智能化设计和制造[12]);(4) 创新平台建设(如:北京材料基因工程高精尖创新中心、上海大学材料基因组工程研究院、西北工业大学材料信息学与基因工程研究中心、哈尔滨工业大学(深圳)材料基因工程及大数据研究院、北京航空航天大学集成计算材料工程中心、云南稀贵金属材料基因工程创新平台[18]);(5) 国内外学术交流(如:材料基因工程高层论坛,创建了专门的刊物:Journal of Materials Informatics和MGE Advances).此外,材料基因工程大大的提高了计算方法和工具与数据库技术在材料学中的地位,由此催生了材料设计的第四范式:数据驱动的材料设计范式[19,20]和科学研究第五范式:人工智能驱动的科学(AI for Science,AI4S)[21]或智能化科研(AI for Research,AI4R)[22]. ...
... 针对上述的问题,刘梓葵教授团队[30]经过在材料热力学与材料动力学领域长期的思考与研究,于近期受邀在Journal of Phase Equilibria and Diffusion上发表了题为Zentropy theory for positive and negative thermal expansion的文章,在文中正式提出了一种通用的计算材料体系多尺度熵的理论框架:叠熵理论(zentropy = z + entropy,其中字母z源于Max Planck为配分函数创造的德语词Zusstandssumme,意为状态总和,也通常用于表示配分函数,该名字是刘梓葵教授在杜克大学讲座时由Josiah Roberts建议的.Zentropy即意为不同尺度/状态熵的叠加之和,因此中文译名为叠熵).该理论一经发表,得到了Phys.org、ScienceDaily、SciTechDaily、AZOMaterials、量子认知百家号等著名科技网站的报道,量子认知百家号给出“综熵理论将是华人科学家们在传统的基础物理理论,特别是热力学基础科学领域作出的重要贡献” (该文中,作者将zentropy译为“综熵”)的高度评价. ...
... [30]Volume-energy curves at 0 K of different configurations<sup>[<xref ref-type="bibr" rid="R30">30</xref>]</sup> (FM, AFM, and NM mean ferromagnetic, antiferromagnetic, and nonmagnetic, respectively; <i>V</i> and <i>E</i><sub>tot</sub> mean atomic volume and total energy at 0 K, respectively)
(a) Ce (b) Fe3Pt ...
... [30] (FM, AFM, and NM mean ferromagnetic, antiferromagnetic, and nonmagnetic, respectively; V and Etot mean atomic volume and total energy at 0 K, respectively)
(a) Ce (b) Fe3Pt ...
Building materials genome from ground‐state configuration to engineering advance
0
2023
Zentropy theory for accurate prediction of free energy, volume, and thermal expansion without fitting parameters
... [45]Temperature-volume phase diagram with isobaric volumes at various pressures<sup>[<xref ref-type="bibr" rid="R45">45</xref>]</sup> (<i>T</i> means temperature; the volume (<i>V</i>) is normalized to their respective equilibrium volume (<i>V</i><sub>N</sub>) at atmospheric pressure and room temperature; CPTE and NTE mean colossal positive thermal expansion and negative thermal expansion, respectively; Exp. means experimental results)
(a) Ce (b) Fe3Pt ...
... [45] (T means temperature; the volume (V) is normalized to their respective equilibrium volume (VN) at atmospheric pressure and room temperature; CPTE and NTE mean colossal positive thermal expansion and negative thermal expansion, respectively; Exp. means experimental results)
(a) Ce (b) Fe3Pt ...
Quantifying the degree of disorder and associated phenomena in materials through zentropy: Illustrated with Invar Fe3Pt
... [46]Predicted degree of disorder (<span class="formulaText"><inline-formula><math id="M36"><msubsup><mrow><mi>f</mi></mrow><mrow><mi mathvariant="normal">D</mi><mi mathvariant="normal">o</mi><mi mathvariant="normal">D</mi></mrow><mrow><mi mathvariant="normal">S</mi></mrow></msubsup></math></span></inline-formula></span><span class="formulaNumber">)</span> as a function of temperature in Fe<sub>3</sub>Pt via entropy<sup>[<xref ref-type="bibr" rid="R46">46</xref>]</sup> (Δ<i>E</i><sub>0</sub> and <i>E<sub>k</sub></i> (<i>V</i>) are equilibrium energy and static total energy at 0 K, respectively. QHA means quasiharmonic approach, in which the free energy with contributions from vibrations and thermal electrons is evaluated)Fig.6<strong>3</strong> 总结与展望
... [46] (ΔE0 and Ek (V) are equilibrium energy and static total energy at 0 K, respectively. QHA means quasiharmonic approach, in which the free energy with contributions from vibrations and thermal electrons is evaluated)Fig.6<strong>3</strong> 总结与展望
... 结合表1中的公式,可计算得到各个组态的概率分布,如图5[47]所示.从图中可以看出,随着温度的升高,铁电基态的概率越来越小,而非基态(尤其是90DW)的概率越来越大,当非基态组态占多数时,即铁电态占比小于50%,可作为铁电-顺电转变的判据,由此得到PbTiO3的铁电-顺电转变温度为776 K (实验结果763 K),实现了完全通过第一性原理无任何经验或者拟合参数的铁电-顺电转变临界温度的预测.此外,考虑到不同微观组态均以一定概率表现,随着温度的升高,PbTiO3中90DW的概率逐渐升高,若铁电态PbTiO3沿晶体c轴极化,则随着温度的增大,将出现大量朝向晶体a轴和b轴方向的极化(与原铁电极化方向形成90DW),在宏观上表现为PbTiO3发生从四方相到立方相的转变.通过该理论能很好地解释X射线衍射(XRD)与X射线吸收精细结构(XAFS)方法(2种方法在时间上的分辨率不同,XRD可以认为是一段时间平均的结果,而XAFS可认为是瞬时的结果)测定PbTiO3宏观和微观晶格参数的矛盾[59]. ...
... [47]Probability of configurations as a function of temperature in PbTiO<sub>3</sub><sup>[<xref ref-type="bibr" rid="R47">47</xref>]</sup> (<i>p</i>—probability, DW—domain wall, FEG—ground state of ferroelectric without DW, <i>T</i><sub>C</sub>—critical temperature, the domain wall energies for 90DW and 180DW are 35 and 132 mJ/m<sup>2</sup>, respectively, which is taken from Ref.[<xref ref-type="bibr" rid="R58">58</xref>])Fig.5<strong>2.4</strong> 无序程度定量描述与有序<strong>-</strong>无序转变预测
... [47] (p—probability, DW—domain wall, FEG—ground state of ferroelectric without DW, TC—critical temperature, the domain wall energies for 90DW and 180DW are 35 and 132 mJ/m2, respectively, which is taken from Ref.[58])Fig.5<strong>2.4</strong> 无序程度定量描述与有序<strong>-</strong>无序转变预测
... 结合表1中的公式,可计算得到各个组态的概率分布,如图5[47]所示.从图中可以看出,随着温度的升高,铁电基态的概率越来越小,而非基态(尤其是90DW)的概率越来越大,当非基态组态占多数时,即铁电态占比小于50%,可作为铁电-顺电转变的判据,由此得到PbTiO3的铁电-顺电转变温度为776 K (实验结果763 K),实现了完全通过第一性原理无任何经验或者拟合参数的铁电-顺电转变临界温度的预测.此外,考虑到不同微观组态均以一定概率表现,随着温度的升高,PbTiO3中90DW的概率逐渐升高,若铁电态PbTiO3沿晶体c轴极化,则随着温度的增大,将出现大量朝向晶体a轴和b轴方向的极化(与原铁电极化方向形成90DW),在宏观上表现为PbTiO3发生从四方相到立方相的转变.通过该理论能很好地解释X射线衍射(XRD)与X射线吸收精细结构(XAFS)方法(2种方法在时间上的分辨率不同,XRD可以认为是一段时间平均的结果,而XAFS可认为是瞬时的结果)测定PbTiO3宏观和微观晶格参数的矛盾[59].
Probability of configurations as a function of temperature in PbTiO<sub>3</sub><sup>[<xref ref-type="bibr" rid="R47">47</xref>]</sup> (<i>p</i>—probability, DW—domain wall, FEG—ground state of ferroelectric without DW, <i>T</i><sub>C</sub>—critical temperature, the domain wall energies for 90DW and 180DW are 35 and 132 mJ/m<sup>2</sup>, respectively, which is taken from Ref.[<xref ref-type="bibr" rid="R58">58</xref>])Fig.5<strong>2.4</strong> 无序程度定量描述与有序<strong>-</strong>无序转变预测
Nature of ferroelectric-paraelectric phase transition and origin of negative thermal expansion in PbTiO3
1
2015
... 结合表1中的公式,可计算得到各个组态的概率分布,如图5[47]所示.从图中可以看出,随着温度的升高,铁电基态的概率越来越小,而非基态(尤其是90DW)的概率越来越大,当非基态组态占多数时,即铁电态占比小于50%,可作为铁电-顺电转变的判据,由此得到PbTiO3的铁电-顺电转变温度为776 K (实验结果763 K),实现了完全通过第一性原理无任何经验或者拟合参数的铁电-顺电转变临界温度的预测.此外,考虑到不同微观组态均以一定概率表现,随着温度的升高,PbTiO3中90DW的概率逐渐升高,若铁电态PbTiO3沿晶体c轴极化,则随着温度的增大,将出现大量朝向晶体a轴和b轴方向的极化(与原铁电极化方向形成90DW),在宏观上表现为PbTiO3发生从四方相到立方相的转变.通过该理论能很好地解释X射线衍射(XRD)与X射线吸收精细结构(XAFS)方法(2种方法在时间上的分辨率不同,XRD可以认为是一段时间平均的结果,而XAFS可认为是瞬时的结果)测定PbTiO3宏观和微观晶格参数的矛盾[59]. ...
Density functional thermodynamic description of spin, phonon and displacement degrees of freedom in antiferromagnetic-to-paramagnetic phase transition in YNiO3






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}