


近年来,人工智能(artificial intelligence,AI)技术突飞猛进,不仅在数据处理和自动化领域取得了显著成绩[1,2],而且在诸多科学领域,如图像识别[3~8]、语音识别[9~11]和自然语言处理[12~14]等,也展示了强大的推动作用。从深度学习的成功运用到计算机视觉和自然语言处理的突破,AI技术已经成为科研创新的一大助力并取得了令人瞩目的成果[15~17]。科学智能化(AI for Science或AI4Science)[18]作为一个新兴的研究方向,正逐渐渗透到科学研究的各个领域,也逐渐成为推动材料研发的新引擎。在材料科学领域,AI技术的引入极大地加速了新材料的发现与设计过程[19~22]。传统材料研发通常依赖于实验和试错法,耗时且成本高昂[23~30]。而深度融合AI的材料研究,使得海量数据驱动的材料性能预测和优化成为可能[31~35]。机器学习技术也被广泛应用于新材料发现、材料性能预测以及生产工艺优化等多个环节[36~42]。基于大量数据训练的机器学习模型,不仅能够预测材料性能[43~47],还能在复杂的化学空间中探索未知的可能性,在结构预测[48~50]、成分设计和合成路径优化[51~57]等方面展现出强大的潜力。
然而,尽管机器学习在提升效率和精度方面展现出了巨大潜力,但这些模型往往被视为“黑盒”,其内部工作机制对于研究人员来说是不透明的[58~60]。由于缺乏对模型决策过程的透明度,研究者往往难以完全理解模型的预测基础,这使得科研人员难以利用这些模型来获取深层次的科学洞见,也难以验证模型预测的准确性和可靠性,因此,机器学习的“黑盒”属性阻碍了材料物理化学知识的探索和技术创新。更进一步,这种不透明性不仅限制了人们对模型预测结果的信任,也阻碍了科学探索的深度与广度[61~63]。因此,如何揭示机器学习的“黑盒”机理成为科学研究的一个难题,探索能够揭示机器学习模型决策过程的可解释机器学习(explainable machine learning,XML)成为材料领域的新焦点,也为材料研发与设计带来了新机遇。
XML旨在打开机器学习模型的“黑盒”,揭示机器学习模型的内部机制,提供对模型决策过程的洞察,增进人们对模型预测背后原因及内在机理的理解[64,65]。进一步而言,对模型的理解意味着研究人员能够了解模型是如何从输入数据中学习并得到目标预测的,哪些特征对目标性能的影响显著,以及模型预测的不确定性来源等。这种解释性不仅有助于提升模型的可信度,还能为研究人员提供新的研究视角和设计思路。在材料领域,XML不仅可帮助研究人员验证模型的有效性,还能增进对材料行为的理解,识别哪些材料特性对性能有重大影响,理解不同变量之间的相互作用,从而加速新材料的发现与创新,有利于在设计新材料或优化现有材料性能时做出更为科学的决策[66~68]。因此,XML在材料研发与设计中至关重要,它不仅是提高机器学习模型透明度和信任度的关键,也是推动材料科学领域持续创新的重要工具。
本文将首先介绍XML的基础知识和发展里程碑;第二部分主要从模型内部结构、不可知方法、局部可解释性和全局可解释性等多个角度探讨XML的分类、常用算法及其在材料研究中的应用;第三部分介绍本团队提出的基于ALKEMIE平台的XML符号回归方法和可视化机器学习方法;最后展望了未来XML在材料学领域的潜在发展方向。
1 XML发展历程
随着技术的不断进步,科学技术的发展从依赖明确算法的时期逐渐步入了基于算法与数据的时代。在传统算法中,问题的解决主要依赖于预设的、透明的算法流程,这些算法为人类提供了清晰的“是什么”“为什么”以及“如何做”的答案,使得整个决策过程具备高度的可解释性[69]。然而,在如今数据驱动的科学时代,机器学习系统通过处理海量数据来学习并生成决策规则,而这些规则往往不再以人类可直接理解的算法形式呈现,从而使得输出结果的内在逻辑变得模糊。在这一转变中,XML的重要性日益凸显。
XML的发展历程是AI这一广泛领域的一个方面,侧重于人类专家可以理解的模型和方法。多年来,这一领域取得了长足的发展,如图1所示。1988年起,研究人员[70~73]开发了ID3和C4.5等算法,提出了最早的决策树和基于规则的可解释系统;Rumelhart等[74]在神经网络中引入了反向传播算法,尽管模型可解释性较低,但其推动了复杂神经网络和深度学习的发展和普及;21世纪以来,随着模型变得越来越复杂,可解释的AI成为热门研究领域[75,76];2016年,Ribeiro等[77]提出了可推测神经网络模型局部可解释性算法(local interpretable model-agnostic explanations,LIME);Lundberg和Lee[78]提出了适用于任意机器学习模型的博弈论算法(shapley additive explanation,SHAP),显著加速了机器学习模型可解释性的研究。2016年后,欧盟立法者和监管机构提出了《通用数据保护条例》(general data protection regulation,GDPR)法规,较早对AI算法的可解释性进行法律规制,主要体现在GDPR第22条,针对完全通过技术方式作出的决策以及不存在人类参与的技术,数据主体的知情权和访问权至少涉及以下3个方面:(1) 告知存在该项处理的事实;(2) 提供关于内在逻辑的有意义信息;(3) 解释该项处理的重要性和预想的后果,且数据主体有权请求人为干预,表达其观点并提出质疑。此项要求将XML可解释性要求纳入GDPR要求,进一步推动了XML的研究。2017年美国信息创新办公室 (Information Innovation Office,I2O)、国防高级研究项目部 (Defense Advanced Research Projects Agency,DARPA)启动了可解释人工智能[79] (explainable artificial intelligence,XAI)项目,并提出了针对XAI的首要目标:构建更加可解释的模型,同时保持高水平的学习性能(预测准确性),使用户能够理解、适当信任并有效管理新一代AI体,如图2a所示。该项目吸引了大量资金和关注度,促进了可解释性机器学习的研究和进步。从图 1中XML的发展历程可以看出,随着法律法规的要求和XAI项目的确立,自2016年来,AI系统在关键领域中的应用越来越频繁,透明度和可解释性的研究进展也呈指数增长,使得加强深度学习模型可解释性成为近期研究的一个重要焦点。
图1
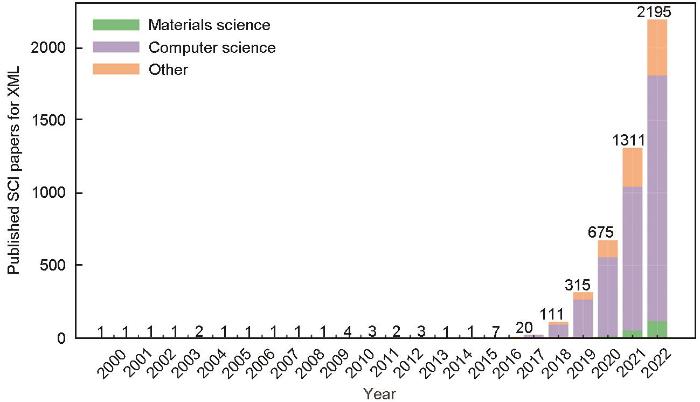
图1
可解释机器学习(XML)发展历程(根据Explainable Machine Learning关键词在Web of Science数据库中检索发表的SCI论文数量)
Fig.1
Development history of explainable machine learning (XML) (based on the number of SCI papers published with the keyword “Explainable Machine Learning” retrieved from the Web of Science database)
图2
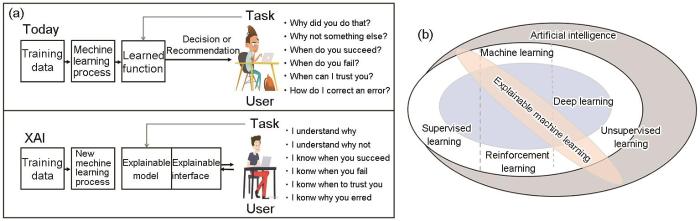
模型的可解释性包含2个核心要素:可理解性和完整性。可理解性指的是能够根据个人需求提供对机器学习模型功能的描述,从而帮助用户获得有意义的知识并对系统建立信任。而完整性则是指模型描述的准确性与其预测结果的可能性紧密相关,一个完整的描述应能全面反映模型的知识和决策逻辑。若对模型的描述能够用人类可理解的语言提取模型的所有知识,则该描述是完整的。然而,可理解性和完整性之间往往是相互制约的,需要进行一定的权衡。高度可解释模型的描述通常较为简单,而追求完整性模型的可解释描述可能与模型本身一样复杂。因此,在实际应用中,需要根据特定的任务目标和应用场景找到这种权衡的最佳点,以确保模型的可解释性既满足用户需求,又保证足够的预测精度和稳定性。
在探索模型的可解释性过程中,要求XML模型本身需要遵循F.A.S.T.原则[69],即:
(1) 公平(Fairness)原则,要求模型在处理数据时不带有任何偏见,避免因数据量增加而产生的固有关联偏差。例如,在自然语言处理中,模型应避免将不相关的单词固定关联起来。
(2) 负责(Accountability)原则,强调模型预测的可靠性,即模型应能对其产生的决策负责,确保预测精度和稳定性。
(3) 安全(Security)原则,要求模型能够抵御恶意攻击,如通过逆向工程利用模型的关键特征进行误导,从而降低模型输出的准确性。可解释模型应能有效消除这类安全隐患。
(4) 透明(Transparency)原则,是指模型内部的工作原理应对人类可理解。这不仅涉及模型决策的过程,还包括其决策的依据和逻辑。例如,在围棋比赛中,AlphaGo的下棋决策虽然高效,但某些步骤如第37手棋的选择对人类来说仍是未解之谜。这种透明度的缺乏正是AI可解释性需要解决的关键问题。
除此之外,探索机器学习模型的可解释性还需要综合考虑以下标准:① 明确性(Clarity),解释必须是清晰明了的,避免使用过于复杂或模糊的语言。解释应该直接针对模型的决策过程,提供具体、直观的信息;② 准确性(Accuracy),提供的解释必须准确地反映模型的决策逻辑。解释不应误导研究人员,而应真实地展现模型是如何处理输入并做出预测的;③ 完整性(Completeness),解释应涵盖模型决策的所有关键因素,不应遗漏任何对决策有重大影响的信息。同时,也要注意完整性和可理解性之间的权衡,避免提供过多细节而导致用户难以理解;④ 一致性(Consistency),对于相似的输入,解释应该是一致的。如果模型的决策逻辑发生变化,解释也应相应地更新以反映这些变化;⑤ 有用性(Usefulness),解释应该为研究人员提供有价值的信息,帮助他们理解模型的决策过程,并能够基于这些信息做出合理的决策;⑥ 可交互性(Interactivity),研究人员在特定情况下可能需要与模型进行交互以获得更深入的解释。因此,XML系统应支持用户提问、提供反馈或调整模型参数等操作。
XML主要有以下3大类应用场景:
(1) 模型验证:判断应用验证模型是否只对特定的数据集使用以及模型是否具有偏差性等,需要查看黑盒模型的构建过程,以评估和判断模型生成决策的标准。在金融风险评估中,XML可以帮助验证模型是否仅针对特定数据集优化,以及是否存在偏差。通过查看黑盒模型的构建过程,评估和判断模型生成决策的标准,确保模型的泛化能力和公正性。在法律评估中,XML可以提供模型决策的法律解释,帮助法官和律师理解基于机器学习决策的背后逻辑。在医疗诊断中,XML可以解释诊断模型的决策过程,增加医生对模型决策的信任,并辅助医生进行更准确的诊断。
(2) 模型调试:通过XML技术,开发人员可以调试机器学习模型,确保其具有高精准性、可靠性和鲁棒性。这包括对输入的微小变化不敏感,并降低模型在面对恶意逆向构建时的风险。当模型出现不当行为或离奇预测时,XML可以帮助定位问题并进行调试,提高模型的性能和稳定性。
(3) 知识发现:XML不仅用于预测,还用于探索对特定过程、事件或系统的理解与认知。在材料科学、生物学、社会学等多个领域,模型需要提供可解释的原因和因果关系,而不仅仅是数值预测。通过XML,研究人员可以增加对未知领域知识的认知,建立更完善的思维模型,并增加人类对模型的信任程度。在没有可解释模型的情况下进行预测被认为是没有科学意义的。
2 XML方法
本部分内容主要从2个维度介绍构建XML模型的常用方法。XML模型可以根据解释的范围划分为2大类:局部可解释性和全局可解释性。局部可解释性侧重于解释模型在特定实例或数据点上的决策过程,它深入剖析了单个预测背后的原因和逻辑;全局可解释性着眼于模型的整体行为和决策规则,提供了对模型在整个数据集上如何运作的全面理解。在另一个维度上,根据是否修改机器学习模型内部结构,将XML方法分为模型内部结构可解释方法和外部评估模型可解释性方法2大类,也是本部分内容介绍的重点。XML方法分类的逻辑图如图3所示。
图3
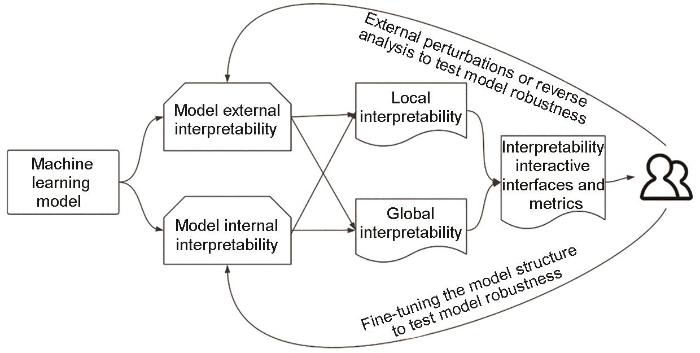
为了直观比较不同XML方法的性能,我们分别从以下6个维度综合考虑可解释模型的性能:
(1) 完整性:评估模型解释覆盖了多大程度的模型行为或决策过程。一个高完整性的解释应能覆盖模型所有重要的决策因素和路径。可以评估不同特征对模型预测的贡献程度,通过逐个移除特征并观察模型表现的变化来评估特征的重要性。
(2) 表达力:用来衡量解释所能表达的信息丰富度。一个表达力强的解释能够提供模型内部运作的详细信息,比如参数、中间计算结果等。提供决策树完整路径的表达力比仅提供特征重要性排名的表达力强,全局解释比局部可解释具有更高的表达力。
(3) 透明性:评估模型的工作原理对于用户的可见程度。一个透明度高的模型可以让用户清楚地看到每一步的计算过程和逻辑。如决策树和线性模型通常被认为比深度神经网络更透明。
(4) 可移植性:评估解释机制能在不同模型或环境中使用的能力。高可移植性的解释方法可以轻松地应用于多种类型的模型。评估解释方法是否依赖于特定的模型类型,以及该解释方法在不同数据集或任务上的适用性。
(5) 算法复杂度:包括时间复杂度(计算时间)和空间复杂度(存储空间)。主要用于评估算法涉及解释生成所需的计算资源和时间。在实际应用中,低复杂度的方法更受欢迎。
(6) 可理解性:模型是否提供了易于理解的输出和解释,以便非专业人士也能够理解模型的预测结果和依据。
2.1 模型内部结构可解释方法
2.1.1 线性回归
线性回归是XML中的一种基础模型,它通过最小化预测值和实际值之间的误差平方和,拟合数据点最佳的直线关系。线性回归的表达式如
式中,
该方法提供对模型行为的直观理解和解释,其特点为:完整性较高,相关系数直接提供线性回归权重值的解释,模型表达力较强;透明性高,可以直接观察模型的内部参数;可移植性较低,解释只依赖线性回归机制;算法复杂度较低,可解释性强,被广泛应用于材料的各个领域。例如:在工程材料数据库中,存在许多关联属性,Doreswamy[80]通过材料数据库分析揭示了聚合物和金属材料屈服强度和抗拉强度等力学属性的相关性,研究结果表明,聚合物材料和金属材料的决定系数(R2)分别为0.9976和0.9994。
2.1.2 逻辑回归
逻辑回归是XML中被广泛使用的一种统计模型[81],主要用于二分类问题。它通过对数几率函数(logit function)将线性回归模型的输出映射到0和1之间,以预测事件的发生概率P。该模型的优势在于其输出的概率解释性,使得模型不仅能预测类别,还能给出预测的不确定性量化。逻辑回归的表达式如
式中,
该方法完整性较高,模型表达力较低,模型系数无法直接理解,透明性较差,不可移植,模型的解释只适用于当前的逻辑回归模型,算法复杂度比线性回归高,可解释性强。
2.1.3 决策树
决策树模型是一种基于树状结构进行决策和预测的监督学习算法[82]。决策树通过递归方法将数据集分割成更小的子集,以树的形式建立分类或回归模型。每个内部节点代表一个属性判断的条件,每个分支代表某个属性的可能取值,每个叶子节点代表一个分类标签或具体数值。决策树易于理解和解释,因为其决策过程可以直观地通过树状图展示出来,每个决策路径都对应一条从根节点到叶子节点的路径,显示了做出特定决策所基于的特征和阈值。决策树模型完整性较高,节点和树叶的模型结构使其具有极高的表达能力;透明性高,可以直接观察模型内部参数;可移植性较高,可以拓展到随机森林和提升树等更复杂的机器学习模型中;算法复杂度中等,可解释性强。在材料领域中,Manna等[83]通过连续的决策树搜索方法开发了混合键序势 (HyBOP),该算法搜索过程如图4[83]所示,被用于预测54种不同族元素的纳米团簇和块体系统的势能面,从而进一步预测材料的能量、原子力、动态稳定性等性质。决策树结合遗传算法等搜索方法,可以构建更加直观的符号回归模型,具体可以参考近期发表的可解释符号回归综述论文[84]。
图4
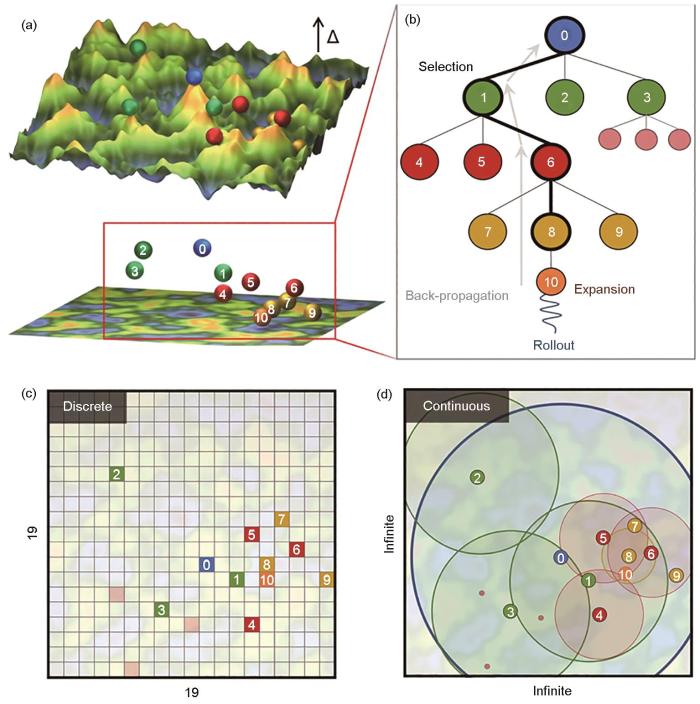
图4
连续决策树搜索算法流程,Monte Carlo树搜索(MCTS)决策树结构中根节点、父节点、子节点及其关系的示意图,传统MCTS算法的搜索空间的离散数据点,及本文中的参数搜索问题[83]
Fig.4
Continuous decision tree search algorithm flow (The spheres represent candidates for different model parameters in the MCTS run, and the numbers on the spheres correspond to their node positions in the Monte Carlo Tree Search (MCTS) tree shown in Fig.4b. These numbers roughly correspond to the exploration order of the candidates, Δ represents the difference between predicted values and objective values) (a); schematic of the root node, parent node, child nodes and their relationships in the MCTS decision tree structure (b); discrete data points in the search space of the traditional MCTS algorithm (c); and parameter search problem in this paper so that the algorithm can converge to the optimal solution (d)[83]
2.1.4 K近邻模型
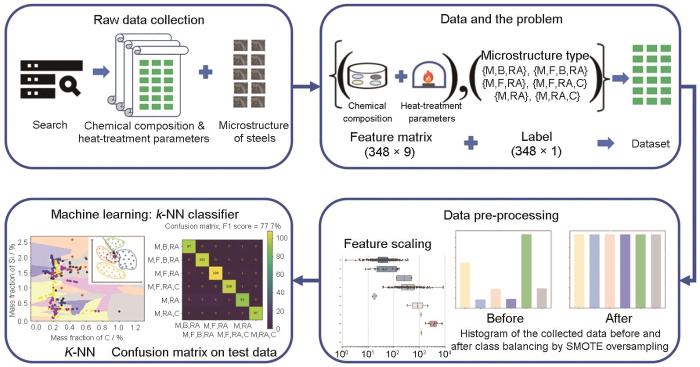
K近邻(K nearest neighbors)算法是一种基于距离度量的分类与回归方法[85]。该算法的核心思想是,一个样本的类别或数值可以由其最近的K个邻居的类别或数值通过投票或平均来确定。算法会计算待分类样本与数据集中每个样本之间的距离,选取距离最近的K个样本,然后根据这些邻居的类别或数值来预测待分类样本的类别或数值。K值的选择、距离度量的方式以及数据的标准化处理等都是影响K近邻性能的关键因素。常用的距离度量方式有Euclidean距离(Euclidean distance)和Manhattan距离(Manhattan distance),而K值是一个自定义的常数,表示在做出预测时考虑的最近邻居的数量。K的选择会影响算法的准确性和复杂度。较小的K值可能使模型更复杂,容易过拟合;较大的K值可能使模型更简单,但可能导致欠拟合。在分类问题中,输入基于其邻居的多数投票分类;在回归问题中,输出是其K个最近邻居的属性的平均值。K近邻算法的主要优点是模型易于理解,无需假设数据分布,使得其在实际应用中非常灵活和强大。Gupta等[86]基于该可解释算法提出了用于对淬火配分钢(quenching-partitioning steel,QPS)显微组织类型进行分类的K近邻构建方法,如图5[86]所示。通过编译包含348个钢样品信息的QPS显微组织类型数据库,研究者利用K近邻算法构建了一个高效的分类模型,模型的特征空间由钢的成分组成、下临界温度(钢在加热过程中开始形成奥氏体的温度Ac1)、上临界温度(钢在加热过程中完全转变为奥氏体的温度Ac3)、马氏体转变温度(Ms)等,以及QPS热处理参数构成。目标变量则记录为显微组织类型,例如马氏体-残留奥氏体{M, RA}、马氏体-贝氏体-残留奥氏体{M, B, RA}等。模型的性能通过F1分数(F1 score)进行评估,该分类器在训练数据集和测试数据集上的预测精度分别达到了97.7%和77.7%,显示出了较高的准确性。基于模型的可解释性,马氏体-残留奥氏体{M, RA}类型被发现是最具混淆性的类别,这也为后续的研究提供了改进方向。
图5
2.2 外部评估模型可解释性方法
2.2.1 特征重要性排序和部分依赖图
特征重要性排序是一种评估各个特征对模型预测性能影响程度的方法[87]。通过分析特征重要性,可以了解哪些特征对模型输出的影响最大,有助于优化特征选择和模型设计。特征重要性的计算方式多样,例如基于树模型的Gini指数或信息增益等,或是通过排列重要性(permutation importance)来评估,即随机排列某个特征的值,观察模型性能的变化,性能下降越多说明该特征越重要。部分依赖图(partial dependence plot,PDP)是一种可视化工具[88],用于展示1个或2个特征对模型预测结果的边际效应。它通过固定其他特征的值,改变感兴趣的特征值,从而观察模型预测结果的变化。部分依赖图有助于理解特征和预测结果之间的复杂关系,如线性关系、单调关系或更复杂的关系。通过部分依赖图,可以直观地看到特征值的变化如何影响模型的预测,进而增强模型的可解释性。由于使用不可知方法实现模型相对可解释性,完整性较低;由于仅关注重要特征,因此模型表达力不强;因为没有深入分析模型参数,所以该方法透明性较低;该方法适用于任何机器学习模型,具有较高的可移植性;模型的复杂度较低,可理解性强。
2.2.2 SHAP加法解释算法
SHAP加法解释算法基于合作博弈论中的Shapley值,为每一个特征分配一个数值,用以表示该特征对模型预测结果的贡献[89]。SHAP算法通过计算每个特征在模型预测中的边际贡献,即该特征存在与否对模型预测结果的影响,来确定每个特征的Shapley值。这个值反映了特征对预测结果的贡献程度,正值表示该特征增加了预测值,而负值则表示该特征降低了预测值。SHAP加法解释算法的优势在于其能够提供一个统一的方法解释任何机器学习模型的预测结果,而且能够考虑到特征之间的相互作用。这使得SHAP算法成为一种非常强大的工具,可以帮助人们更好地理解机器学习模型的决策过程,从而提高模型的透明度和可解释性。对于集合(N)中的每个元素i,其Shapley值
式中,
(1) TreeSHAP算法[90],专门针对树模型(如决策树、随机森林、XGBoost等)的SHAP值计算方法,它利用了树模型的结构特点,通过递归的方法计算每个节点上的SHAP值高效地得到每个特征的贡献,计算效率非常高。
(2) KernelSHAP[91]方法,其适用于无法使用上述特定模型去解释的复杂模型,如深度学习模型或某些集成学习方法。它使用核方法(kernel method)来估计SHAP值。核方法允许对模型进行黑箱处理,不依赖于模型的具体形式,只关注模型的输入和输出。这使得KernelSHAP具有广泛的适用性。但是由于KernelSHAP需要对每个数据点进行多次模型评估,因此可能需要大量的计算资源。
(3) DeepSHAP[92]方法,是针对深度学习模型的一种SHAP值计算方法。由于深度学习模型的复杂性,直接应用KernelSHAP可能会非常耗时。DeepSHAP通过结合深度学习模型的特性和SHAP值的理论,提供了一种更高效的计算方式。
2.2.3 层级相关性传播方法
层级相关性传播(layer-wise relevance propagation,LRP)方法是一种解释深度学习模型预测结果的方法[95]。它的核心目标是理解模型对输入的预测是如何形成的,以及每个输入特征对最终预测结果的贡献。LRP方法基于反向传播,在传播过程中引入了权重的概念,旨在量化每个神经元对下一层神经元的贡献,如图6蓝色线条即为当前神经元对下一层神经元的贡献度。LRP方法计算出每个输入特征对模型输出的具体影响,通过特定的规则将每个神经元的“重要性”或“相关性”传播回前一层。这些规则根据神经元的激活度和连接权重进行调整,以确保前一层的神经元根据其对输出的贡献获得适当的“重要性”。LRP方法不仅适用于特定的深度学习模型,而且还可以广泛应用于各种神经网络结构,包括卷积神经网络和循环神经网络等。其解释结果可以生成与输入数据具有相同矩阵大小和维度的像素级热图,从而可视化了输入图像中对所选类别有贡献的重要区域,直观地展示了输入特征的相对重要性,从而帮助研究者理解模型的决策过程。由于LRP方法在材料领域鲜有应用,因此本文以生物蛋白质中每个原子对解离常数的可解释预测为例,为LRP方法在探究材料实验和计算模型的可解释性应用方面提供指导。Cho等[96]将图神经网络和LRP可解释性技术相结合,用于模拟蛋白质和配体之间非共价相互作用,与共价相互作用相比,非共价相互作用由于其连接模糊性受到的关注较少。作者以蛋白质数据银行(protein data bank,PDB)中存储的一个特定的三维结构3F7H蛋白质-配体复合物为例,其三维结构如图7a[96]所示,基于生物学知识的蛋白质-配体相互作用如图7b[96]所示,其中NH末端的氢键用红色虚线表示,疏水相互作用用灰色虚线表示。通过LRP分析,该模型成功捕获了给定复合物中蛋白质和配体之间的重要非共价相互作用。PotentialNet和InteractionNet 2个模型中每个原子对预测结果的贡献热力图如图7c和d[96]所示,InteractionNet模型相比于PotentialNet模型具有更高的准确性,并且识别出了容易形成H键的NH原子对解离常数预测的积极贡献,表明蛋白质中的解离常数与复合物中实际的H键有很强的相关性。此外,LRP方法已经被广泛地应用于医学癌症诊断、人体睡眠阶段自动可解释分类等问题中。
图6
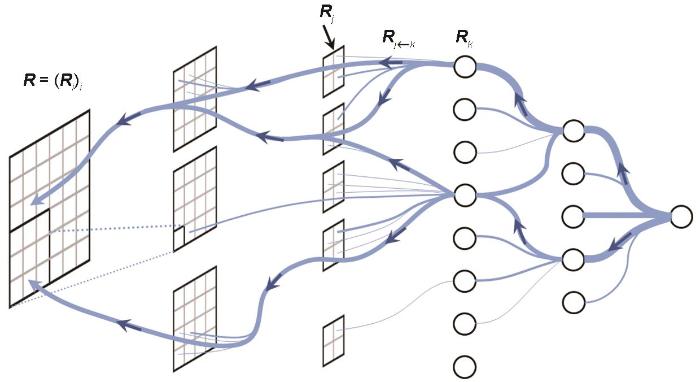
图6
层级相关性传播(LRP)方法中的反向传播示意图
Fig.6
Schematic of backpropagation in layer-wise relevance propagation (LRP) method ( Ri —relevance score of the input feature, Rj —relevance score of the feature in an intermediate layer, Rk —relevance score of the feature in the output layer,
图7
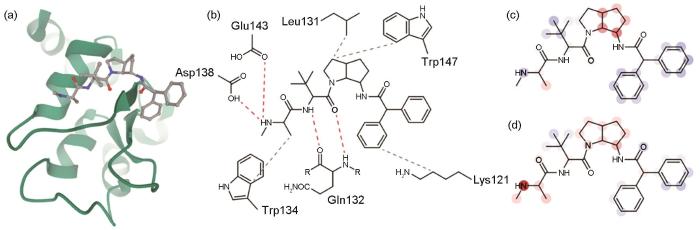
图7
蛋白质-配体复合物3F7H的三维结构,基于生物学知识的蛋白质-配体相互作用,及PotentialNet模型和InteractionNet模型通过LRP方法获得的原子对解离常数预测贡献的热图[96]
Fig.7
Three-dimensional structure of the protein-ligand complex, 3F7H (The protein is depicted in a cartoon (green), and the ligand is depicted in color-coded ball-and-stick. Atom colors: gray (C), red (O), and blue (N)) (a); knowledge-based estimation of protein-ligand interactions (Hydrogen bonds are depicted in red dashed lines, and hydrophobic contacts are depicted in gray dashed lines) (b); heat map for the atomic contributions on the prediction of interaction, obtained from the LRP on PotentialNet (c); and heat map for the atomic contributions on the prediction of interaction, obtained from the LRP on InteractionNet (d) (The contributions are illustrated with color intensity of red (positive influence), white (zero influence), and blue (negative influence) colors)[96]
2.2.4 类别激活映射图(class activation maps,CAM)
类别激活映射图是一种可视化技术,用于解释深度学习模型,特别是卷积神经网络在分类任务中的决策过程[97]。CAM通过高亮显示图像中对于模型预测某一类别起到关键作用的区域,从而提供直观的可视化效果。它基于模型的最后一个卷积层的特征图和全局平均池化层的权重来生成。首先,模型对输入矩阵数据进行前向传播,获取最后一个卷积层的特征图。然后,这些特征图通过全局平均池化层转换为特征向量,并与分类层的权重相乘。乘积的结果被用来生成CAM,图上每个位置的值表示该位置对模型预测某一类别的贡献度。CAM可以帮助研究人员理解模型是如何根据输入图像的特定区域做出预测的。通过观察CAM,可以识别出图像中对于分类结果最重要的部分,从而提高模型的可解释性。基于CAM技术的可视化机器学习方法目前在材料学中暂时没有应用,但是Luo等[98]基于CAM思想,构建了可以识别和分类的透射电子(TEM)像中碳纳米管/纳米纤维(carbon nanotubes/nanofibers,CNT/CNF)颗粒的机器学习模型并探究了不同模型所识别到的关键特征,如图8[98]所示。空气中的碳质纳米材料很可能形成单个纳米颗粒和微米级团聚体的混合物,其结构复杂、形状不规则,使得结构识别和分类极其困难。虽然TEM像的手动分类被广泛使用,但由于缺乏用于结构识别的自动化工具,这种方法非常耗时。Luo等[98]基于卷积神经网络(网络结构如图8i[98]所示),结合迁移学习和K近邻聚类等方法,在8类数据集(图8a~h[98])上实现了84.5%的准确率,并给出了该分类模型在每一类数据中识别到的关键数据点,为理解模型如何实现TEM像的自动分类依据提供了直观的可解释分析。CAM提供了一种直观的方式来理解模型的决策过程,无需修改模型结构或重新训练。但是CAM的局限性在于它只能提供模型对于特定类别的决策依据,而不能解释模型的整体行为。
图8
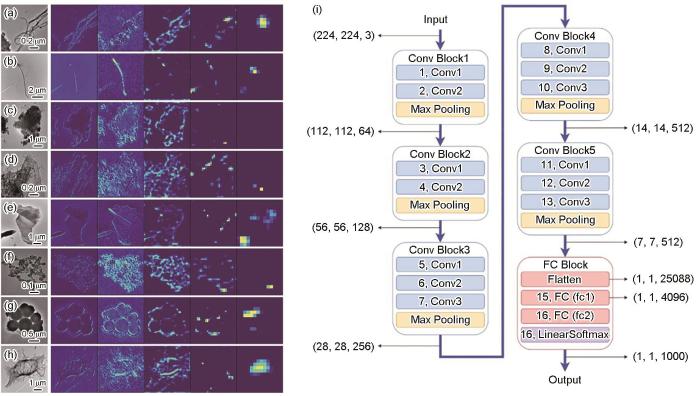
图8
类别激活映射图(CAM)在图片分类问题中的示意图[98]
Fig.8
Examples of hypercolumn representations for eight image classes: cluster (a), fiber (b), matrix (c), matrix-surface (d), hraphene sheets (e), soot particles (f), high-density particles (g), and polymer residuals (h); Schematic of class activation map (CAM) in image classification problem[98] (Conv Block—convolutional neural network unit. FC Block—fully connected unit) (i)
2.2.5 其他深度神经网络可解释方法
针对深度神经网络模型的可解释性方法,除了上述4种方法外,还有基于遮挡(perturbation-based)的方法和神经元响应最大化(activation maximization)等方法。
基于遮挡的方法旨在通过观察和分析模型在不同输入扰动下的输出从而解释模型的决策过程[99]。通过对输入数据进行局部遮挡或扰动,观察模型预测的变化,从而推断出输入特征对模型输出的影响。首先,选择一个或多个需要解释的数据点;然后,对这些数据点的不同部分进行遮挡或扰动;其次,将扰动后的数据输入到模型中,并记录模型的预测结果;最后,通过对比分析不同扰动下的预测结果,可以推断出哪些特征对模型的决策起到了重要作用。基于遮挡的方法适用于各种机器学习模型,尤其是深度学习模型。它可以帮助研究人员理解模型在处理图像、文本等复杂数据类型时的决策依据。该方法的优点在于直观性和灵活性,可以通过简单的遮挡操作来探索模型对不同输入的敏感性。然而,其局限性在于可能无法全面反映模型内部的复杂决策过程,且对于大规模数据集和高维数据可能效率较低。
神经元响应最大化旨在直观展示深度学习模型中特定神经元学到的特征[100]。该技术通过寻找能够使特定神经元激活值最大化的输入模式,来理解该神经元所学习到的特征。即找到能最大程度激活某个神经元的输入数据。该方法通常涉及对网络输入进行迭代优化,以最大化选定神经元的激活值。优化过程可以使用梯度上升等优化算法,通过计算神经元激活关于输入的梯度,并逐步调整输入以最大化激活。一旦找到最大化神经元激活的输入,就可以将其可视化,从而直观地理解神经元所代表的特征。这对于解释深度学习模型的内部工作原理非常有帮助。虽然神经元响应最大化可以提供有价值的直观解释,但优化过程可能受到初始值、优化算法和超参数选择的影响,可能引起不同的最大化输入等问题。
2.3 XML方法总结
针对上述XML方法,本文将所有模型对应的完整性、表达力、透明性、可移植性、算法复杂度、可理解性能力汇总如表1所示。
表 1 XML方法评估结果
Table 1
| Classification | XML Method | Integrity | Expressiveness | Transparency | Portability | Complexity | Understandability |
|---|---|---|---|---|---|---|---|
Interpretable methods from internal model structure | Linear regression Logistic regression | High | High | High | Low | Simple | Easy |
| High | Lowa | Low | Not portable | Middle | Easy | ||
| Decision tree | High | High | High | Highb | Middle | Easy | |
| K nearest neighbors | High | High | High | Not portable | Simple | Easy | |
| Interpretable methods for external model evaluation methods | Feature importance ranking and partial dependence plots | Low | Limitedc | Low | Highd | Simple | Easy |
| Shapley additive explanation(SHAP) | Lowe | Extremely high for single predictions | Low | High | Middle | Easy | |
| LRP | Low | High | High | High | Complex | Easy | |
| CAM | Low | High | High | High | Complex | Easy |
目前常用的XML软件包括:
(1) LIME,最早提出的可解释性方法之一,用于解释机器学习模型的预测。它可以帮助理解模型学习的内容和为什么以某种方式进行预测。代码库:
(2) Shapash,包含可视化模型解释的Python库,代码库:
(3) InterpretML,支持训练可解释模型和解释现有的黑盒机器学习模型,代码库:
(4) ELI5 (explain like I am fine),调试和解释机器学习分类器的预测。它支持多个机器学习框架,如scikit-learn、XGBoost、LightGBM、CatBoost和Keras等。代码库:
(5) OmniXAI,提供全面可解释的AI和可解释的机器学习能力,旨在解释各种类型的数据、模型和解释技术,为数据科学家和机器学习研究人员提供一站式的综合库。代码库:
(6) SHAP,一种基于博弈论方法开发的pythonXML库,用于解释任何机器学习模型的输出。SHAP利用经典Shapley值及其相关扩展将最优方案与局部解释联系起来,代码库:
(7) XAITK (explainable AI toolkit),该工具包是根据DARPA's XAI计划开发的,包含DARPA项目开发的多种XML方法的工具和资源,可帮助用户、开发人员和研究人员理解复杂的机器学习模型,代码库:
(8) MatterSR,基于符号回归的XML模型构建方法,主要用于基于决策树和随机森林构建具有数学方程的函数表达式,我们将在下文ALKEMIE平台部分详细论述,目前该方法已在催化材料、高熵合金材料等领域获得了应用,代码库:
3 ALKEMIE 智能计算平台的XML
3.1 基于ALKEMIE云平台的可解释符号回归方法
本团队基于ALKEMIE Cloud[101]和ALKEMIE Matter Studio[102]平台,开发了XML符号回归方法,并在MBene材料单原子催化剂的析氧反应(oxygen evolution reaction,OER)和氧还原反应(oxygen reduction reaction,ORR)性能预测上获得了广泛应用。Wang等[103]首先通过密度泛函理论计算获得了包含120个初始数据点的二维过渡金属硼化物(MBene)材料OER和ORR的催化性能数据集,进一步将其划分为训练集、测试集和验证集。对于材料描述符或材料特征,选择了39个与材料组成和结构相关的信息,并通过XML方法中的2.2.1小节中的特征重要性排序和部分依赖图去除了高度重复特征,保留了10个最重要的独立特征。分别建立了2个针对OER和ORR性能的机器学习模型,分别实现了高达97%和98%的预测准确率。进一步地,为了探究OER和ORR催化性能与催化原子和基体结构的数学表达关系,结合随机森林和遗传算法构建了符号回归XML方法,如图9[103]所示。该方法针对OER和ORR催化性能的线性相关系数分别为0.79和0.88,均方根误差分别为0.094和0.095,预测精度分别为75.36%和79.91%。尽管符号回归方法相比黑盒模型性能有所损失,但是该方法给出了OER和ORR性能与其重要特征间的数学表达式,分别如
式中,
图9
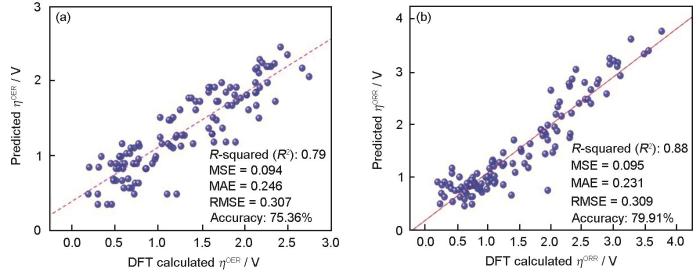
图9
基于可解释符号回归方法的二维过渡金属硼化物(MBene)单原子催化剂析氧反应(OER)和氧还原反应(ORR)活性预测模型[103]
Fig.9
Prediction model for oxygen evolution reaction (OER) (a) and oxygen reduction reaction (ORR) (b) reactivity of transition metal boride (MBene) single-atom catalysts based on interpretable symbolic regression (ηORR and ηOER—efficiencies of ORR or OER, respectively; MSE—squared error; MAE—mean absolute error; RMSE—root mean squared error; DFT—density functional theory)[103]
这些表达式不仅提供了对OER和ORR效率的新见解,而且为探索材料特征之间的物理化学规律提供了新思路。
3.2 ALKEMIE 平台可视化机器学习方法
可交互和可视化的机器学习平台在XML领域中起着至关重要的作用,ALKEMIE多尺度高通量计算与数据管理智能平台构建了针对线性回归、逻辑回归、决策树和K近邻的可视化和可交互软件平台[104~109],如图10所示。该平台不仅推动了该领域的技术进步,也促进了其在实际应用中的广泛应用。该平台通过直观的可视化界面,使得复杂的机器学习模型变得易于理解和操作,可使非专业用户也能够轻松参与到模型的开发和优化过程中。同时,平台提供的交互式功能可使用户能够实时查看模型训练过程中的各种指标和参数变化,从而更深入地理解模型的工作原理和性能表现。随着数据科学和AI技术的不断发展,可交互和可视化的机器学习平台将继续在XML领域中发挥重要作用,助力更多领域实现智能化升级。
图10
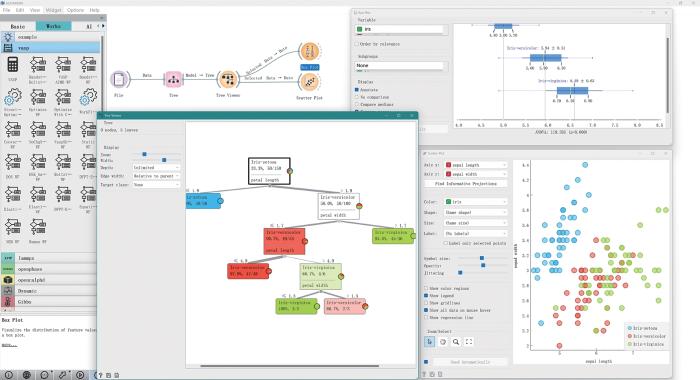
图10
ALKEMIE 多尺度高通量计算与数据管理智能平台中的可视化机器学习
Fig.10
Visualized machine learning in ALKEMIE multi-scale high-throughput computing and data management intelligent platform
4 总结与展望
本论文主要介绍了可解释机器学习方法的基础概念,常用的可解释机器学习方法以及在材料研发中的关键应用和未来展望。首先概述了可解释机器学习方法发展历程及重要的里程碑,以及可解释机器学习在人工智能领域中的定位和需要遵守的F.A.S.T.原则;第二部分根据是否修改机器学习模型内部结构,分别介绍了包括线性回归、逻辑回归、决策树和K近邻算法等模型内部结构可解释方法,特征重要性排序、部分依赖图、SHAP加法解释算法、逐层相关性分数传播、CAM、基于遮挡和神经元响应最大化等方法的外部评估模型可解释性方法;第三部分介绍了本团队ALKEMIE多尺度高通量计算与数据管理智能平台中的可解释符号回归算法和可视化机器学习;最后,展望了可解释机器学习模型未来发展的研究方向。
尽管XML模型在材料学科研究中取得了一定的成果,但仍存在诸多问题:
(1) 目前XML在材料学中仅针对模型内部结构可解释方法有少量应用,而材料中基于深度学习的绝大多数模型不具备可解释性。因此,未来如何探究针对材料性能预测或分类问题中深度学习模型的可解释性,结合生成式预训练大模型(generative pre-trained transformer,GPT)等海量基础知识,探究深度模型或符号回归表达式背后隐藏的物理或化学意义是未来重点研究方法之一。
(2) 模型的可解释性与精度之间往往存在一定的权衡。增加模型的可解释性可能会牺牲部分预测精度,如何构建兼具模型精度和可解释性的方法至关重要。
(3) 当前的XML技术还未能完全满足材料科学家对于模型内部工作机制的理解需求,尤其是在处理复杂的非线性关系时。随着材料数据的不断积累和更新,如何有效地整合域知识以提高模型的泛化能力和解释性,并确保模型在持续学习过程中的可解释性也是一个重要研究方向。
参考文献
Machine learning: Trends, perspectives, and prospects
[J].
Machine learning addresses the question of how to build computers that improve automatically through experience. It is one of today's most rapidly growing technical fields, lying at the intersection of computer science and statistics, and at the core of artificial intelligence and data science. Recent progress in machine learning has been driven both by the development of new learning algorithms and theory and by the ongoing explosion in the availability of online data and low-cost computation. The adoption of data-intensive machine-learning methods can be found throughout science, technology and commerce, leading to more evidence-based decision-making across many walks of life, including health care, manufacturing, education, financial modeling, policing, and marketing. Copyright © 2015, American Association for the Advancement of Science.
Computational vision and regularization theory
[A].
Going deeper with convolutions
[A].
Deep residual learning for image recognition
[A].
You only look once: Unified, real-time object detection
[A].
ImageNet classification with deep convolutional neural networks
[J].
Deep learning for computer vision: A brief review
[J].
Machine learning paradigms for speech recognition: An overview
[J].
Deep audio-visual speech recognition
[J].
PANNs: Large-scale pretrained audio neural networks for audio pattern recognition
[J].
Jumping NLP curves: A review of natural language processing research
[J].
Attention is all you need
[A].
Advances, challenges and opportunities in creating data for trustworthy AI
[J].
Convolutional networks for images, speech, and time series
[A].
Deep learning
[J].
Learning deep generative models
[J].
AI for science: Report on the department of energy (DOE) town halls on artificial intelligence (AI) for science
[R].
Machine-learning-assisted materials discovery using failed experiments
[J].
Data mining uncovers a treasure trove of topological materials
[J].
Machine learning-aided engineering of hydrolases for pet depolymerization
[J].
Machine learning-enabled high-entropy alloy discovery
[J].High-entropy alloys are solid solutions of multiple principal elements that are capable of reaching composition and property regimes inaccessible for dilute materials. Discovering those with valuable properties, however, too often relies on serendipity, because thermodynamic alloy design rules alone often fail in high-dimensional composition spaces. We propose an active learning strategy to accelerate the design of high-entropy Invar alloys in a practically infinite compositional space based on very sparse data. Our approach works as a closed-loop, integrating machine learning with density-functional theory, thermodynamic calculations, and experiments. After processing and characterizing 17 new alloys out of millions of possible compositions, we identified two high-entropy Invar alloys with extremely low thermal expansion coefficients around 2 × 10 per degree kelvin at 300 kelvin. We believe this to be a suitable pathway for the fast and automated discovery of high-entropy alloys with optimal thermal, magnetic, and electrical properties.
The high-throughput highway to computational materials design
[J].High-throughput computational materials design is an emerging area of materials science. By combining advanced thermodynamic and electronic-structure methods with intelligent data mining and database construction, and exploiting the power of current supercomputer architectures, scientists generate, manage and analyse enormous data repositories for the discovery of novel materials. In this Review we provide a current snapshot of this rapidly evolving field, and highlight the challenges and opportunities that lie ahead.
High-throughput electronic band structure calculations: Challenges and tools
[J].
Advances in data‐assisted high‐throughput computations for material design
[J].
A combinatorial approach to materials discovery
[J].A method that combines thin film deposition and physical masking techniques has been used for the parallel synthesis of spatially addressable libraries of solid-state materials. Arrays containing different combinations, stoichiometries, and deposition sequences of BaCO(3), Bi(2)O(3), CaO, CuO, PbO, SrCO(3), and Y(2)O(3) were generated with a series of binary masks. The arrays were sintered and BiSrCaCuO and YBaCuO superconducting films were identified. Samples as small as 200 micrometers by 200 micrometers in size were generated, corresponding to library densities of 10,000 sites per square inch. The ability to generate and screen combinatorial libraries of solid-state compounds, when coupled with theory and empirical observations, may significantly increase the rate at which novel electronic, magnetic, and optical materials are discovered and theoretical predictions tested.
Thermal conductivity imaging at micrometre-scale resolution for combinatorial studies of materials
[J].Combinatorial methods offer an efficient approach for the development of new materials. Methods for generating combinatorial samples of materials, and methods for characterizing local composition and structure by electron microprobe analysis and electron-backscatter diffraction are relatively well developed. But a key component for combinatorial studies of materials is high-spatial-resolution measurements of the property of interest, for example, the magnetic, optical, electrical, mechanical or thermal properties of each phase, composition or processing condition. Advances in the experimental methods used for mapping these properties will have a significant impact on materials science and engineering. Here we show how time-domain thermoreflectance can be used to image the thermal conductivity of the cross-section of a Nb-Ti-Cr-Si diffusion multiple, and thereby demonstrate rapid and quantitative measurements of thermal transport properties for combinatorial studies of materials. The lateral spatial resolution of the technique is 3.4 microm, and the time required to measure a 100 x 100 pixel image is approximately 1 h. The thermal conductivity of TiCr(2) decreases by a factor of two in crossing from the near-stoichiometric side of the phase to the Ti-rich side; and the conductivity of (Ti,Nb)(3)Si shows a strong dependence on crystalline orientation.
Data‐driven materials innovation and applications
[J].
Progress in materials genome engineering in China
[J].Materials genome engineering (MGE) is a frontier technology in the field of material science and engineering, which is well capable to revolutionize the research and development (R&D) mode of new materials, greatly improve the R&D efficiency, shorten the R&D time, and reduce the cost. This paper reviews the progress of MGE in China from the aspects of the fundamental theory and methods, key technology and equipment, the R&D of new materials and related engineering application, talents training, formation and promotion of new concept of material genetic engineering. The paper also looks forward to the future development of MGE in China.
中国材料基因工程研究进展
[J].材料基因工程是材料领域的颠覆性前沿技术,将对材料研发模式产生革命性的变革,全面加速材料从设计到工程化应用的进程,大幅度提升新材料的研发效率,缩短研发周期,降低研发成本,促进工程化应用。本文从基础理论与方法、关键技术与装备、新材料研发与工程化应用、人才培养以及材料基因工程新理念的形成和推广等方面,综述了中国材料基因工程的研究进展,并提出了未来发展方向建议。
Toward new-generation intelligent manufacturing
[J].
Materials data infrastructure: A case study of the Citrination platform to examine data import, storage, and access
[J].
Big data creates new opportunities for materials research: A review on methods and applications of machine learning for materials design
[J].
View and comments on the data ecosystem: “Ocean of data”
[J].
Materials data toward machine learning: Advances and challenges
[J].
Dramatically enhanced combination of ultimate tensile strength and electric conductivity of alloys via machine learning screening
[J].
Machine learning assisted composition effective design for precipitation strengthened copper alloys
[J].
Machine learning for materials research and development
[J].The rapid advancement of big data and artificial intelligence has resulted in new data-driven materials research and development (R&D), which has achieved substantial progress. This fourth paradigm is believed to improve materials design efficiency and industrialized application and stimulate the discovery of new materials. The focus of this work is on the emerging field of machine learning-assisted material R&D, with an emphasis on machine learning predictions and optimization design. Following a brief description of feature construction and selection, recent developments in material predictions on phases/structures, processing-structure-property relationships, microstructure, and material performance are reviewed. This paper also summarizes the research progress on optimization algorithms with machine learning models, which is expected to overcome the bottlenecks such as the small size and high noise level of material data samples and huge space for exploration. The challenges and future opportunities for machine learning applications in materials R&D are discussed and prospected.
机器学习在材料研发中的应用
[J].大数据和人工智能技术的快速发展推动数据驱动的材料研发快速发展成为变革传统试错法的新模式,即所谓的材料研发第四范式。新模式将大幅度提升材料研发效率和工程化应用水平,推动新材料快速发展。本文聚焦机器学习辅助材料研发这一新兴领域,以材料预测和优化设计为主线,在简述材料特征构建与筛选的基础上,综述了机器学习在材料相结构、显微组织、成分-工艺-性能、服役行为预测等方面的研究进展;针对材料数据样本量少、噪音高、质量差,以及新材料探索空间巨大的特点,综述了机器学习模型与优化算法和策略融合,在新材料优化设计中的研究进展和典型应用。最后,讨论了机器学习在材料领域的发展机遇和挑战,展望了发展前景。
Symbolic regression in materials science via dimension-synchronous-computation
[J].There is growing interest in applying machine learning techniques in the field of materials science. However, the interpretation and knowledge extracted from machine learning models is a major concern, particularly as formulating an explicit model that provides insight into physics is the goal of learning. In the present study, we propose a framework that utilizes the filtering ability of feature engineering, in conjunction with symbolic regression to extract explicit, quantitative expressions for the band gap energy from materials data. We propose enhancements to genetic programming with dimensional consistency and artificial constraints to improve the search efficiency of symbolic regression. We show how two descriptors attributed to volumetric and electronic factors, from 32 possible candidates, explicitly express the band gap energy of NaCl-type compounds. Our approach provides a basis to capture underlying physical relationships between materials descriptors and target properties.
General-purpose machine-learned potential for 16 elemental metals and their alloys
[J].
Transition metal and n doping on alp monolayers for bifunctional oxygen electrocatalysts: Density functional theory study assisted by machine learning description
[J].
Hydrogen-promoted heterogeneous plastic strain and associated hardening effect in polycrystalline nickel under uniaxial tension
[J].
Distilling free-form natural laws from experimental data
[J].For centuries, scientists have attempted to identify and document analytical laws that underlie physical phenomena in nature. Despite the prevalence of computing power, the process of finding natural laws and their corresponding equations has resisted automation. A key challenge to finding analytic relations automatically is defining algorithmically what makes a correlation in observed data important and insightful. We propose a principle for the identification of nontriviality. We demonstrated this approach by automatically searching motion-tracking data captured from various physical systems, ranging from simple harmonic oscillators to chaotic double-pendula. Without any prior knowledge about physics, kinematics, or geometry, the algorithm discovered Hamiltonians, Lagrangians, and other laws of geometric and momentum conservation. The discovery rate accelerated as laws found for simpler systems were used to bootstrap explanations for more complex systems, gradually uncovering the "alphabet" used to describe those systems.
Interaction trends between single metal atoms and oxide supports identified with density functional theory and statistical learning
[J].
Machine learning of mechanical properties of steels
[J].
Simple descriptor derived from symbolic regression accelerating the discovery of new perovskite catalysts
[J].Symbolic regression (SR) is an approach of interpretable machine learning for building mathematical formulas that best fit certain datasets. In this work, SR is used to guide the design of new oxide perovskite catalysts with improved oxygen evolution reaction (OER) activities. A simple descriptor, μ/t, where μ and t are the octahedral and tolerance factors, respectively, is identified, which accelerates the discovery of a series of new oxide perovskite catalysts with improved OER activity. We successfully synthesise five new oxide perovskites and characterise their OER activities. Remarkably, four of them, CsLaMnCoO, CsLaNiO, SrNiCoO, and SrBaNiO, are among the oxide perovskite catalysts with the highest intrinsic activities. Our results demonstrate the potential of SR for accelerating the data-driven design and discovery of new materials with improved properties.
Hierarchical symbolic regression for identifying key physical parameters correlated with bulk properties of perovskites
[J].
Inverse molecular design using machine learning: Generative models for matter engineering
[J].The discovery of new materials can bring enormous societal and technological progress. In this context, exploring completely the large space of potential materials is computationally intractable. Here, we review methods for achieving inverse design, which aims to discover tailored materials from the starting point of a particular desired functionality. Recent advances from the rapidly growing field of artificial intelligence, mostly from the subfield of machine learning, have resulted in a fertile exchange of ideas, where approaches to inverse molecular design are being proposed and employed at a rapid pace. Among these, deep generative models have been applied to numerous classes of materials: rational design of prospective drugs, synthetic routes to organic compounds, and optimization of photovoltaics and redox flow batteries, as well as a variety of other solid-state materials.Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
A property-oriented design strategy for high performance copper alloys via machine learning
[J].
Physics guided deep learning for generative design of crystal materials with symmetry constraints
[J].
Virtual screening of inorganic materials synthesis parameters with deep learning
[J].
Planning chemical syntheses with deep neural networks and symbolic AI
[J].
Network analysis of synthesizable materials discovery
[J].Assessing the synthesizability of inorganic materials is a grand challenge for accelerating their discovery using computations. Synthesis of a material is a complex process that depends not only on its thermodynamic stability with respect to others, but also on factors from kinetics, to advances in synthesis techniques, to the availability of precursors. This complexity makes the development of a general theory or first-principles approach to synthesizability currently impractical. Here we show how an alternative pathway to predicting synthesizability emerges from the dynamics of the materials stability network: a scale-free network constructed by combining the convex free-energy surface of inorganic materials computed by high-throughput density functional theory and their experimental discovery timelines extracted from citations. The time-evolution of the underlying network properties allows us to use machine-learning to predict the likelihood that hypothetical, computer-generated materials will be amenable to successful experimental synthesis.
Bayesian reaction optimization as a tool for chemical synthesis
[J].
Can we predict materials that can be synthesised?
[J].
Nanoparticle synthesis assisted by machine learning
[J].
Machine-learning a solution for reactive atomistic simulations of energetic materials
[J].
Practical black-box attacks against machine learning
[A].
Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead
[J].Black box machine learning models are currently being used for high stakes decision-making throughout society, causing problems throughout healthcare, criminal justice, and in other domains. People have hoped that creating methods for explaining these black box models will alleviate some of these problems, but trying to black box models, rather than creating models that are in the first place, is likely to perpetuate bad practices and can potentially cause catastrophic harm to society. There is a way forward - it is to design models that are inherently interpretable. This manuscript clarifies the chasm between explaining black boxes and using inherently interpretable models, outlines several key reasons why explainable black boxes should be avoided in high-stakes decisions, identifies challenges to interpretable machine learning, and provides several example applications where interpretable models could potentially replace black box models in criminal justice, healthcare, and computer vision.
Transferability in machine learning: From phenomena to black-box attacks using adversarial samples
[J].
Understanding black-box predictions via influence functions
[A].
Opening the black box: The promise and limitations of explainable machine learning in cardiology
[J].
A survey on the explainability of supervised machine learning
[J].
Physics-informed neural networks: A deep learning framework for solving forward and inverse problems involving nonlinear partial differential equations
[J].We introduce physics-informed neural networks - neural networks that are trained to solve supervised learning tasks while respecting any given laws of physics described by general nonlinear partial differential equations. In this work, we present our developments in the context of solving two main classes of problems: data-driven solution and data-driven discovery of partial differential equations. Depending on the nature and arrangement of the available data, we devise two distinct types of algorithms, namely continuous time and discrete time models. The first type of models forms a new family of data-efficient spatio-temporal function approximators, while the latter type allows the use of arbitrarily accurate implicit Runge-Kutta time stepping schemes with unlimited number of stages. The effectiveness of the proposed framework is demonstrated through a collection of classical problems in fluids, quantum mechanics, reaction-diffusion systems, and the propagation of nonlinear shallow-water waves. (C) 2018 Elsevier Inc.
Unsupervised word embeddings capture latent knowledge from materials science literature
[J].
Physics-informed machine learning
[J].The rapidly developing field of physics-informed learning integrates data and mathematical models seamlessly, enabling accurate inference of realistic and high-dimensional multiphysics problems. This Review discusses the methodology and provides diverse examples and an outlook for further developments.
ID5: An incremental ID3
[A].
Incremental induction of decision trees
[J].
Improved use of continuous attributes in C4.5
[J].
A comparative study of decision tree ID3 and C4.5
[J].
Learning representations by back-propagating errors
[J].
Explainable machine learning for scientific insights and discoveries
[J].
Accelerating the design of compositionally complex materials via physics-informed artificial intelligence
[J].The chemical space for designing materials is practically infinite. This makes disruptive progress by traditional physics-based modeling alone challenging. Yet, training data for identifying composition-structure-property relations by artificial intelligence are sparse. We discuss opportunities to discover new chemically complex materials by hybrid methods where physics laws are combined with artificial intelligence.© 2023. Springer Nature America, Inc.
“Why should I trust you?” Explaining the predictions of any classifier
[A].
A unified approach to interpreting model predictions
[A].
DARPA's explainable AI (XAI) program: A retrospective
[J].
Linear regression model for knowledge discovery in engineering materials
[A].
Decision tree methods: Applications for classification and prediction
[J].
Learning in continuous action space for developing high dimensional potential energy models
[J].Reinforcement learning (RL) approaches that combine a tree search with deep learning have found remarkable success in searching exorbitantly large, albeit discrete action spaces, as in chess, Shogi and Go. Many real-world materials discovery and design applications, however, involve multi-dimensional search problems and learning domains that have continuous action spaces. Exploring high-dimensional potential energy models of materials is an example. Traditionally, these searches are time consuming (often several years for a single bulk system) and driven by human intuition and/or expertise and more recently by global/local optimization searches that have issues with convergence and/or do not scale well with the search dimensionality. Here, in a departure from discrete action and other gradient-based approaches, we introduce a RL strategy based on decision trees that incorporates modified rewards for improved exploration, efficient sampling during playouts and a "window scaling scheme" for enhanced exploitation, to enable efficient and scalable search for continuous action space problems. Using high-dimensional artificial landscapes and control RL problems, we successfully benchmark our approach against popular global optimization schemes and state of the art policy gradient methods, respectively. We demonstrate its efficacy to parameterize potential models (physics based and high-dimensional neural networks) for 54 different elemental systems across the periodic table as well as alloys. We analyze error trends across different elements in the latent space and trace their origin to elemental structural diversity and the smoothness of the element energy surface. Broadly, our RL strategy will be applicable to many other physical science problems involving search over continuous action spaces.© 2022. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Exploring the mathematic equations behind the materials science data using interpretable symbolic regression
[J].
A machine learning model for multi-class classification of quenched and partitioned steel microstructure type by the k-nearest neighbor algorithm
[J].
Classification with correlated features: Unreliability of feature ranking and solutions
[J].Classification and feature selection of genomics or transcriptomics data is often hampered by the large number of features as compared with the small number of samples available. Moreover, features represented by probes that either have similar molecular functions (gene expression analysis) or genomic locations (DNA copy number analysis) are highly correlated. Classical model selection methods such as penalized logistic regression or random forest become unstable in the presence of high feature correlations. Sophisticated penalties such as group Lasso or fused Lasso can force the models to assign similar weights to correlated features and thus improve model stability and interpretability. In this article, we show that the measures of feature relevance corresponding to the above-mentioned methods are biased such that the weights of the features belonging to groups of correlated features decrease as the sizes of the groups increase, which leads to incorrect model interpretation and misleading feature ranking.With simulation experiments, we demonstrate that Lasso logistic regression, fused support vector machine, group Lasso and random forest models suffer from correlation bias. Using simulations, we show that two related methods for group selection based on feature clustering can be used for correcting the correlation bias. These techniques also improve the stability and the accuracy of the baseline models. We apply all methods investigated to a breast cancer and a bladder cancer arrayCGH dataset and in order to identify copy number aberrations predictive of tumor phenotype.R code can be found at: http://www.mpi-inf.mpg.de/~laura/Clustering.r.
PDP: An R package for constructing partial dependence plots
[J].
Failure mode and effects analysis of RC members based on machine-learning-based Shapley additive explanations (SHAP) approach
[J].
Fast TreeSHAP: Accelerating SHAP value computation for trees
[J].
Improving KernelSHAP: Practical Shapley value estimation using linear regression
[A].
Explaining a series of models by propagating Shapley values
[J].Local feature attribution methods are increasingly used to explain complex machine learning models. However, current methods are limited because they are extremely expensive to compute or are not capable of explaining a distributed series of models where each model is owned by a separate institution. The latter is particularly important because it often arises in finance where explanations are mandated. Here, we present Generalized DeepSHAP (G-DeepSHAP), a tractable method to propagate local feature attributions through complex series of models based on a connection to the Shapley value. We evaluate G-DeepSHAP across biological, health, and financial datasets to show that it provides equally salient explanations an order of magnitude faster than existing model-agnostic attribution techniques and demonstrate its use in an important distributed series of models setting.© 2022. The Author(s).
SHAP-IQ: Unified approximation of any-order Shapley interactions
[A].
Model interpretability of financial fraud detection by group SHAP
[J].
Layer-wise relevance propagation: An overview
[A].
Layer-wise relevance propagation of interactionnet explains protein-ligand interactions at the atom level
[J].Development of deep-learning models for intermolecular noncovalent (NC) interactions between proteins and ligands has great potential in the chemical and pharmaceutical tasks, including structure-activity relationship and drug design. It still remains an open question how to convert the three-dimensional, structural information of a protein-ligand complex into a graph representation in the graph neural networks (GNNs). It is also difficult to know whether a trained GNN model learns the NC interactions properly. Herein, we propose a GNN architecture that learns two distinct graphs-one for the intramolecular covalent bonds in a protein and a ligand, and the other for the intermolecular NC interactions between the protein and the ligand-separately by the corresponding covalent and NC convolutional layers. The graph separation has some advantages, such as independent evaluation on the contribution of each convolutional step to the prediction of dissociation constants, and facile analysis of graph-building strategies for the NC interactions. In addition to its prediction performance that is comparable to that of a state-of-the art model, the analysis with an explainability strategy of layer-wise relevance propagation shows that our model successfully predicts the important characteristics of the NC interactions, especially in the aspect of hydrogen bonding, in the chemical interpretation of protein-ligand binding.
LayerCAM: Exploring hierarchical class activation maps for localization
[J].
A transfer learning approach for improved classification of carbon nanomaterials from tem images
[J].The extensive use of carbon nanomaterials such as carbon nanotubes/nanofibers (CNTs/CNFs) in industrial settings has raised concerns over the potential health risks associated with occupational exposure to these materials. These exposures are commonly in the form of CNT/CNF-containing aerosols, resulting in a need for a reliable structure classification protocol to perform meaningful exposure assessments. However, airborne carbonaceous nanomaterials are very likely to form mixtures of individual nano-sized particles and micron-sized agglomerates with complex structures and irregular shapes, making structure identification and classification extremely difficult. While manual classification from transmission electron microscopy (TEM) images is widely used, it is time-consuming due to the lack of automation tools for structure identification. In the present study, we applied a convolutional neural network (CNN) based machine learning and computer vision method to recognize and classify airborne CNT/CNF particles from TEM images. We introduced a transfer learning approach to represent images by hypercolumn vectors, which were clustered -means and processed into a Vector of Locally Aggregated Descriptors (VLAD) representation to train a softmax classifier with the gradient boosting algorithm. This method achieved 90.9% accuracy on the classification of a 4-class dataset and 84.5% accuracy on a more complex 8-class dataset. The developed model established a framework to automatically detect and classify complex carbon nanostructures with potential applications that extend to the automated structural classification for other nanomaterials.This journal is © The Royal Society of Chemistry.
Perturbation-based methods for explaining deep neural networks: A survey
[J].
Activation maximization generative adversarial nets
[J].
High-throughput automatic integrated material calculations and data management intelligent platform and the application in novel alloys
[J].
高通量自动流程集成计算与数据管理智能平台及其在合金设计中的应用
[J].材料研发模式经历了经验主导的第一范式、理论模型主导的第二范式和计算模拟主导的第三范式,如今正处于数据驱动的第四范式。为加速新材料的设计与研发,发展基于材料数据库和人工智能算法的高通量自动集成计算和数据挖掘算法变得至关重要。本文介绍了作者团队自主开发的分布式高通量自动流程集成计算和数据管理智能平台ALKEMIE2.0 (Artificial Learning and Knowledge Enhanced Materials Informatics Engineering 2.0),该平台基于AMDIV设计理念,包含了自动化、模块化、数据库、人工智能和可视化流程等5个适用于数据驱动的材料研发模式核心要素。概括来说,ALKEMIE2.0以模块化的方式集成了多个不同尺度的计算模拟软件;其高通量自动纠错流程可实现从建模、运行到数据分析,全程自动无人工干预;支持单用户不低于10<sup>4</sup>量级的并发高通量自动计算模拟。进一步而言,ALKEMIE2.0具有强大的可移植性和可扩展性,目前已部署在国家超算天津中心,基于多类型材料数据库结合超算强大的计算能力使得人工智能算法在新材料设计与研发中得以快速的应用和实践。更重要的是,ALKEMIE2.0设计了用户友好的可视化操作界面,使得结构建模、工作流计算逻辑、数据分析和机器学习模型具有更高的透明性和更强的可操作性,且适用于对材料计算模拟掌握程度从初级到专业的所有材料研究人员。最后,通过多平台部署和高通量筛选二元铝合金2个算例详细展示了ALKEMIE2.0的主要特色及功能。
ALKEMIE: An intelligent computational platform for accelerating materials discovery and design
[J].
MBenes-supported single atom catalysts for oxygen reduction and oxygen evolution reaction by first-principles study and machine learning
[J].
Role of carbon-rings in polycrystalline GeSb2Te4 phase-change material
[J].
Prediction of thermoelectric performance for layered IV-V-VI semiconductors by high-throughput ab initio calculations and machine learning
[J].
PotentialMind: Graph convolutional machine learning potential for Sb-Te binary compounds of multiple stoichiometries
[J].
Machine learning interatomic potential: Bridge the gap between small-scale models and realistic device-scale simulations
[J].
Atomic insights into device-scale phase-change memory materials using machine learning potential
[J].
Accelerating the discovery of transition metal borides by machine learning on small data sets
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}