Ultrahigh specific strength in a magnesium alloy strengthened by spinodal decomposition
1
2021
... 镁合金因其质量轻、资源丰富以及比强度/比刚度高而广泛应用于航空航天、汽车等领域[1~3].Mg-Sn合金成本低,具有反萤石结构的Mg2Sn沉淀相的沉淀硬化效应显著[4~6],也可以改善合金抗蠕变性能[7,8].但Mg2Sn相会快速长大并粗化,从而恶化Mg-Sn合金的力学性能. ...
Phase transformations in an ultralight BCC Mg alloy during anisothermal ageing
0
2022
A near row matching approach to prediction of multiple precipitation crystallography of compound precipitates and its application to a Mg/Mg2Sn system
1
2017
... 镁合金因其质量轻、资源丰富以及比强度/比刚度高而广泛应用于航空航天、汽车等领域[1~3].Mg-Sn合金成本低,具有反萤石结构的Mg2Sn沉淀相的沉淀硬化效应显著[4~6],也可以改善合金抗蠕变性能[7,8].但Mg2Sn相会快速长大并粗化,从而恶化Mg-Sn合金的力学性能. ...
Sequential precipitation behavior of Mg17Al12 and Mg2Sn in Mg-8Al-2Sn-1Zn alloys
2
2018
... 镁合金因其质量轻、资源丰富以及比强度/比刚度高而广泛应用于航空航天、汽车等领域[1~3].Mg-Sn合金成本低,具有反萤石结构的Mg2Sn沉淀相的沉淀硬化效应显著[4~6],也可以改善合金抗蠕变性能[7,8].但Mg2Sn相会快速长大并粗化,从而恶化Mg-Sn合金的力学性能. ...
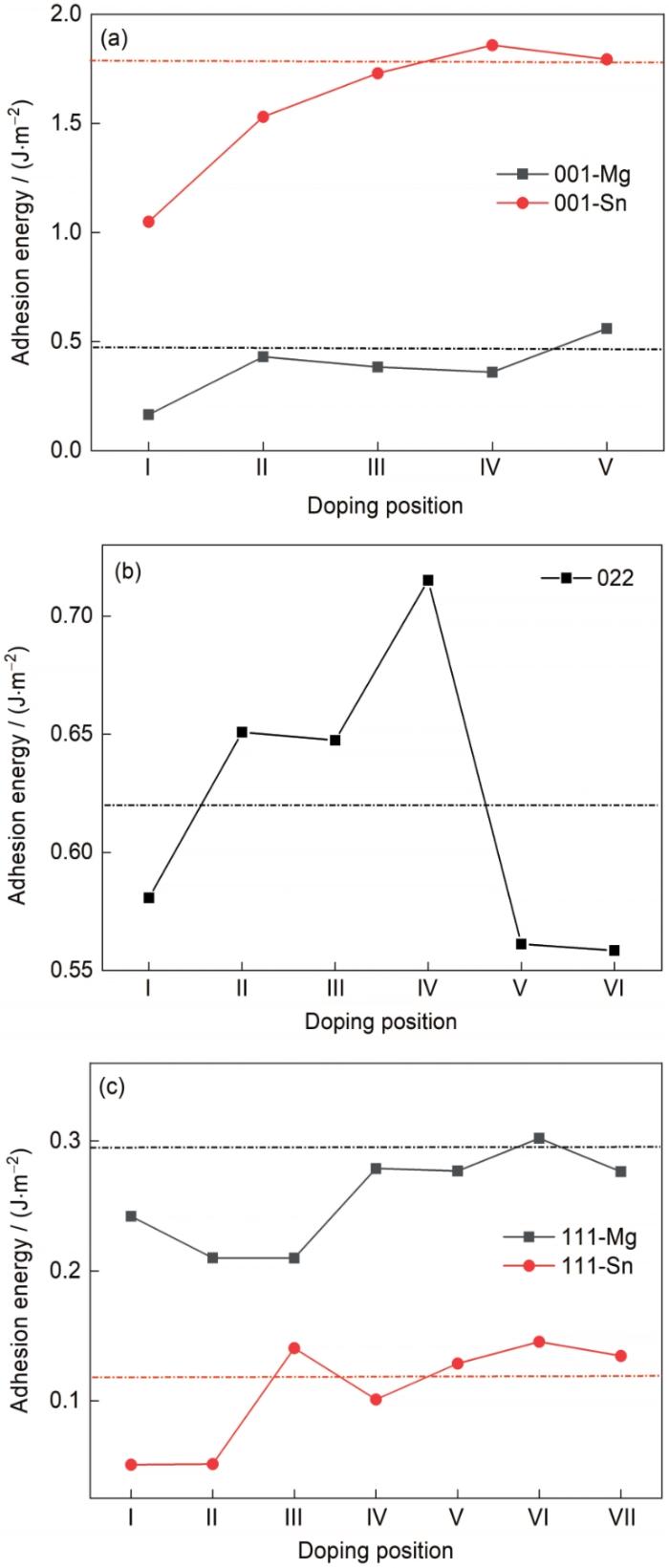
... Mg/Mg2Sn界面结构基于hcp结构的Mg基体[30]以及fcc结构的Mg2Sn析出相[31]建立.对于Mg基体结构,晶格常数a = b = 0.320 nm,c = 0.513 nm;α = β = 90°,γ = 120° (α、β和γ分别为晶格常数a和b、a和c、b和c 3个轴之间的夹角).Mg2Sn析出相为fcc结构,a = b = c = 0.681 nm,α = β = γ = 90°.有实验观察到Mg2Sn与Mg基体之间的取向关系为//Mg(0001)取向(200℃左右)[12]和Mg(0001)//Mg2Sn(022)[4],但考虑到不同取向界面能不一样,本工作分别对Mg(0001)以及Mg2Sn 3个低指数面组成的界面进行第一性原理计算.经过收敛性测试,采用4层Mg2Sn和4层Mg构建了Mg(0001)/Mg2Sn(001)Mg端和Sn端界面、Mg(0001)/Mg2Sn(022)界面以及Mg(0001)/Mg2Sn(111) Mg端和Sn端界面,对其界面间距进行收敛性测试,并测量其界面能和晶格错配度,如表1所示. ...
Microstructure and mechanical properties of binary Mg-Sn alloys subjected to indirect extrusion
0
2010
Suppression of discontinuous precipitation in AZ91 by addition of Sn
1
2014
... 镁合金因其质量轻、资源丰富以及比强度/比刚度高而广泛应用于航空航天、汽车等领域[1~3].Mg-Sn合金成本低,具有反萤石结构的Mg2Sn沉淀相的沉淀硬化效应显著[4~6],也可以改善合金抗蠕变性能[7,8].但Mg2Sn相会快速长大并粗化,从而恶化Mg-Sn合金的力学性能. ...
Effect of Sn on the formation of the long period stacking ordered phase and mechanical properties of Mg-RE-Zn alloy
1
2014
... 镁合金因其质量轻、资源丰富以及比强度/比刚度高而广泛应用于航空航天、汽车等领域[1~3].Mg-Sn合金成本低,具有反萤石结构的Mg2Sn沉淀相的沉淀硬化效应显著[4~6],也可以改善合金抗蠕变性能[7,8].但Mg2Sn相会快速长大并粗化,从而恶化Mg-Sn合金的力学性能. ...
First-principle study on structural stability of Sn alloying MgZn2 phase and Mg2Sn phase
1
2010
... 镁合金因其质量轻、资源丰富以及比强度/比刚度高而广泛应用于航空航天、汽车等领域[1~3].Mg-Sn合金成本低,具有反萤石结构的Mg2Sn沉淀相的沉淀硬化效应显著[4~6],也可以改善合金抗蠕变性能[7,8].但Mg2Sn相会快速长大并粗化,从而恶化Mg-Sn合金的力学性能. ...
Sn合金化MgZn2相及Mg2Sn相结构稳定性的第一原理研究
1
2010
... 镁合金因其质量轻、资源丰富以及比强度/比刚度高而广泛应用于航空航天、汽车等领域[1~3].Mg-Sn合金成本低,具有反萤石结构的Mg2Sn沉淀相的沉淀硬化效应显著[4~6],也可以改善合金抗蠕变性能[7,8].但Mg2Sn相会快速长大并粗化,从而恶化Mg-Sn合金的力学性能. ...
Zn segregation in interface between Mg17Al12 precipitate and Mg matrix in Mg-Al-Zn alloys
1
2019
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
Compositional optimization of Mg-Sn-Al alloys for higher age hardening response
1
2013
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
Enhanced age hardening response by the addition of Zn in Mg-Sn alloys
1
2006
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
Refinement of precipitate distributions in an age-hardenable Mg-Sn alloy through microalloying
2
2006
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
... Mg/Mg2Sn界面结构基于hcp结构的Mg基体[30]以及fcc结构的Mg2Sn析出相[31]建立.对于Mg基体结构,晶格常数a = b = 0.320 nm,c = 0.513 nm;α = β = 90°,γ = 120° (α、β和γ分别为晶格常数a和b、a和c、b和c 3个轴之间的夹角).Mg2Sn析出相为fcc结构,a = b = c = 0.681 nm,α = β = γ = 90°.有实验观察到Mg2Sn与Mg基体之间的取向关系为//Mg(0001)取向(200℃左右)[12]和Mg(0001)//Mg2Sn(022)[4],但考虑到不同取向界面能不一样,本工作分别对Mg(0001)以及Mg2Sn 3个低指数面组成的界面进行第一性原理计算.经过收敛性测试,采用4层Mg2Sn和4层Mg构建了Mg(0001)/Mg2Sn(001)Mg端和Sn端界面、Mg(0001)/Mg2Sn(022)界面以及Mg(0001)/Mg2Sn(111) Mg端和Sn端界面,对其界面间距进行收敛性测试,并测量其界面能和晶格错配度,如表1所示. ...
Enhancing mechanical properties of Mg-Sn alloys by combining addition of Ca and Zn
1
2015
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
Microstructure and mechanical properties of as-cast Mg-Sn-Al-Zn alloy
2
2014
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
... 在实验研究中,Al元素是改善镁合金力学性能的主要元素之一.有不少研究证实了随着Mg-Sn合金中Mg2Sn的不断析出,Mg2Sn相会粗化,导致合金力学性能变差.添加Al元素可以抑制合金内Mg2Sn析出相的粗化,对Mg2Sn起到细化作用,提高合金的力学性能[14~16].然而实验上只是在宏观上分析了Al的添加改善了Mg-Sn界面的力学性能,相比于实验上只是观测到Al掺杂有益于提高Mg/Mg2Sn界面的稳定性,本工作从原子尺度分析解释了掺杂Al元素对Mg/Mg2Sn 3种不同取向界面稳定性的影响,以及界面处不同掺杂位置对界面性质的影响,发现掺杂位置会影响Mg/Mg2Sn界面的稳定性,有的掺杂位置对提高界面的稳定性并不是有益的.Al掺杂后的Mg(0001)/Mg2Sn(022)界面能降低了0.07 eV/nm,这与徐孝新[17]在实验上观察到在Mg-Sn合金中加入Al元素后Al偏聚在Mg基体与Mg2Sn界面,导致界面能降低的结果一致.并且通过对Al掺杂Mg(0001)/Mg2Sn(022)界面电子特性的分析,界面处Al—Sn键强度占主导地位,解释了实验上观测到的Al元素的加入增强了Mg-Sn界面的结合强度. ...
铸态Mg-Sn-Al-Zn合金组织和力学性能
2
2014
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
... 在实验研究中,Al元素是改善镁合金力学性能的主要元素之一.有不少研究证实了随着Mg-Sn合金中Mg2Sn的不断析出,Mg2Sn相会粗化,导致合金力学性能变差.添加Al元素可以抑制合金内Mg2Sn析出相的粗化,对Mg2Sn起到细化作用,提高合金的力学性能[14~16].然而实验上只是在宏观上分析了Al的添加改善了Mg-Sn界面的力学性能,相比于实验上只是观测到Al掺杂有益于提高Mg/Mg2Sn界面的稳定性,本工作从原子尺度分析解释了掺杂Al元素对Mg/Mg2Sn 3种不同取向界面稳定性的影响,以及界面处不同掺杂位置对界面性质的影响,发现掺杂位置会影响Mg/Mg2Sn界面的稳定性,有的掺杂位置对提高界面的稳定性并不是有益的.Al掺杂后的Mg(0001)/Mg2Sn(022)界面能降低了0.07 eV/nm,这与徐孝新[17]在实验上观察到在Mg-Sn合金中加入Al元素后Al偏聚在Mg基体与Mg2Sn界面,导致界面能降低的结果一致.并且通过对Al掺杂Mg(0001)/Mg2Sn(022)界面电子特性的分析,界面处Al—Sn键强度占主导地位,解释了实验上观测到的Al元素的加入增强了Mg-Sn界面的结合强度. ...
Underlying mechanisms of drastic reduction in yield asymmetry of extruded Mg-Sn-Zn alloy by Al addition
1
2018
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
Effects of Zn/Al mass ratio on microstructure evolution and mechanical properties of Mg-Sn based alloys
2
2021
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
... 在实验研究中,Al元素是改善镁合金力学性能的主要元素之一.有不少研究证实了随着Mg-Sn合金中Mg2Sn的不断析出,Mg2Sn相会粗化,导致合金力学性能变差.添加Al元素可以抑制合金内Mg2Sn析出相的粗化,对Mg2Sn起到细化作用,提高合金的力学性能[14~16].然而实验上只是在宏观上分析了Al的添加改善了Mg-Sn界面的力学性能,相比于实验上只是观测到Al掺杂有益于提高Mg/Mg2Sn界面的稳定性,本工作从原子尺度分析解释了掺杂Al元素对Mg/Mg2Sn 3种不同取向界面稳定性的影响,以及界面处不同掺杂位置对界面性质的影响,发现掺杂位置会影响Mg/Mg2Sn界面的稳定性,有的掺杂位置对提高界面的稳定性并不是有益的.Al掺杂后的Mg(0001)/Mg2Sn(022)界面能降低了0.07 eV/nm,这与徐孝新[17]在实验上观察到在Mg-Sn合金中加入Al元素后Al偏聚在Mg基体与Mg2Sn界面,导致界面能降低的结果一致.并且通过对Al掺杂Mg(0001)/Mg2Sn(022)界面电子特性的分析,界面处Al—Sn键强度占主导地位,解释了实验上观测到的Al元素的加入增强了Mg-Sn界面的结合强度. ...
Microstructural characterization of aged Mg-Sn-Al alloy
2
2019
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
... 在实验研究中,Al元素是改善镁合金力学性能的主要元素之一.有不少研究证实了随着Mg-Sn合金中Mg2Sn的不断析出,Mg2Sn相会粗化,导致合金力学性能变差.添加Al元素可以抑制合金内Mg2Sn析出相的粗化,对Mg2Sn起到细化作用,提高合金的力学性能[14~16].然而实验上只是在宏观上分析了Al的添加改善了Mg-Sn界面的力学性能,相比于实验上只是观测到Al掺杂有益于提高Mg/Mg2Sn界面的稳定性,本工作从原子尺度分析解释了掺杂Al元素对Mg/Mg2Sn 3种不同取向界面稳定性的影响,以及界面处不同掺杂位置对界面性质的影响,发现掺杂位置会影响Mg/Mg2Sn界面的稳定性,有的掺杂位置对提高界面的稳定性并不是有益的.Al掺杂后的Mg(0001)/Mg2Sn(022)界面能降低了0.07 eV/nm,这与徐孝新[17]在实验上观察到在Mg-Sn合金中加入Al元素后Al偏聚在Mg基体与Mg2Sn界面,导致界面能降低的结果一致.并且通过对Al掺杂Mg(0001)/Mg2Sn(022)界面电子特性的分析,界面处Al—Sn键强度占主导地位,解释了实验上观测到的Al元素的加入增强了Mg-Sn界面的结合强度. ...
时效Mg-Sn-Al合金的微观结构研究
2
2019
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
... 在实验研究中,Al元素是改善镁合金力学性能的主要元素之一.有不少研究证实了随着Mg-Sn合金中Mg2Sn的不断析出,Mg2Sn相会粗化,导致合金力学性能变差.添加Al元素可以抑制合金内Mg2Sn析出相的粗化,对Mg2Sn起到细化作用,提高合金的力学性能[14~16].然而实验上只是在宏观上分析了Al的添加改善了Mg-Sn界面的力学性能,相比于实验上只是观测到Al掺杂有益于提高Mg/Mg2Sn界面的稳定性,本工作从原子尺度分析解释了掺杂Al元素对Mg/Mg2Sn 3种不同取向界面稳定性的影响,以及界面处不同掺杂位置对界面性质的影响,发现掺杂位置会影响Mg/Mg2Sn界面的稳定性,有的掺杂位置对提高界面的稳定性并不是有益的.Al掺杂后的Mg(0001)/Mg2Sn(022)界面能降低了0.07 eV/nm,这与徐孝新[17]在实验上观察到在Mg-Sn合金中加入Al元素后Al偏聚在Mg基体与Mg2Sn界面,导致界面能降低的结果一致.并且通过对Al掺杂Mg(0001)/Mg2Sn(022)界面电子特性的分析,界面处Al—Sn键强度占主导地位,解释了实验上观测到的Al元素的加入增强了Mg-Sn界面的结合强度. ...
First-principle study on Mg2 X (X = Si, Ge, Sn) intermetallics by Bi micro-alloying
1
2021
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
First-principle studies on the mechanical, thermodynamic and electronic properties of β″-Mg3Gdand β'-Mg7Gd alloys under pressure
0
2019
High-throughput computing for hydrogen transport properties in ε-ZrH2
1
2022
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
From classical thermodynamics to phase-field method
1
2022
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
First-principles study of fluorine-doped zinc oxide
0
2010
Insight into the strengthening mechanism of α-Al2O3/γ-Fe ceramic-metal interface doped with Cr, Ni, Mg, and Ti
0
2021
Exploring failure mode and enhancement mechanism of doped rare-earth elements iron-based/alumina-ceramic interface
1
2022
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
First principle study on alloying effect of Al and Zn doping on Mg-Li phase interface
1
2014
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
Al、Zn对Mg-Li相界合金化效应的第一性原理研究
1
2014
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
Interface structure characterization and elements doping on interface bonding strength and tensile failure mechanism of NiCo coating/Cu matrix
1
2021
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
First-principles investigation on stability and electronic structure of Sc-doped θ'/Al interface in Al-Cu alloys
1
2021
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
Stability of phase boundary between L12-Ni3Al phases: A phase field study
1
2022
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
Role of interfacial energy anisotropy in dendrite orientation in Al-Zn alloys: A phase field study
1
2022
... 通过微合金化可以减少Mg2Sn的析出从而提高合金的性能,这得益于其固溶强化效应[9,10].Sasaki等[11]研究发现,添加Zn元素可以改变Mg2Sn和Mg基体之间的界面能,有利于细化Mg2Sn沉淀物.Mendis等[12]将Mg2Sn作为模型,分别添加Na和In + Li,发现添加Na使得硬化增量比添加In + Li增加了近一倍.Pan等[13]研究证实添加Ca可以在Mg-Sn合金中形成热稳定相CaMgSn,之后添加Y和Zr可以细化CaMgSn相,从而提高合金强度和延展性.田树科和郭学锋[14]发现在Mg-Sn合金中分别添加Al和Zn元素时,都可以提高合金的固溶强化效果,Al的固溶强化效果高于Zn.Kim和Park[15]发现Al元素的加入可以改善Mg-Sn合金的力学性能和硬度.Luo等[16]证实了Al的添加可以显著改变析出相Mg2Sn的分布和形貌,并对Mg2Sn起到细化作用,改善Mg-Sn合金的力学性能.徐孝新[17]发现在Mg-Sn合金中加入Al元素后,Al主要偏聚在Mg基体与Mg2Sn界面处,降低Mg2Sn的界面能,减小形核功,使形核过程更易于进行.然而实验上难以从微观角度分析界面结合机制,需要结合第一性原理[18~20]、分子动力学等方法进行分析.目前第一性原理在研究合金化元素改善界面性质方面取得了很多成果[21~24].王小宏等[25]基于第一性原理研究了Al、Zn占位对Mg/Li相界断裂强度的影响,表明Al、Zn元素的添加提高了体系稳定性.Liu等[26]预测了Cr、Os、Ir元素掺杂对NiCo/Cu界面的结合强度和力学性能的影响,并通过原子位置、键长和电子性质解释了加强机理.Zhang等[27]通过第一性原理发现了Sc在S1位点掺杂AlCu/Al界面会降低其界面能,并且增强其黏附功,特别是被间隙Cu原子占据的S1位点与Sc的掺杂具有非常好的结合强度.在Mg-Sn合金中,粗化的Mg2Sn相会导致该合金的时效硬化效果降低,而Al元素可以显著提高Mg-Sn合金的时效硬化效应.在实验中观测到在Mg-Sn合金中添加Al元素后,Al元素主要偏聚在Mg基体与Mg2Sn相的界面,起到细化作用.第一性原理可以定性给出界面处的电子结构以及合金元素对界面稳定性[28,29]的影响,但对于Al掺杂Mg/Mg2Sn界面性质的研究很少. ...
Interfacial segregation and fracture in Mg-based binary alloys: Experimental and first-principles perspective
1
2018
... Mg/Mg2Sn界面结构基于hcp结构的Mg基体[30]以及fcc结构的Mg2Sn析出相[31]建立.对于Mg基体结构,晶格常数a = b = 0.320 nm,c = 0.513 nm;α = β = 90°,γ = 120° (α、β和γ分别为晶格常数a和b、a和c、b和c 3个轴之间的夹角).Mg2Sn析出相为fcc结构,a = b = c = 0.681 nm,α = β = γ = 90°.有实验观察到Mg2Sn与Mg基体之间的取向关系为//Mg(0001)取向(200℃左右)[12]和Mg(0001)//Mg2Sn(022)[4],但考虑到不同取向界面能不一样,本工作分别对Mg(0001)以及Mg2Sn 3个低指数面组成的界面进行第一性原理计算.经过收敛性测试,采用4层Mg2Sn和4层Mg构建了Mg(0001)/Mg2Sn(001)Mg端和Sn端界面、Mg(0001)/Mg2Sn(022)界面以及Mg(0001)/Mg2Sn(111) Mg端和Sn端界面,对其界面间距进行收敛性测试,并测量其界面能和晶格错配度,如表1所示. ...
Mg2Sn: A potential mid-temperature thermoelectric material
1
2016
... Mg/Mg2Sn界面结构基于hcp结构的Mg基体[30]以及fcc结构的Mg2Sn析出相[31]建立.对于Mg基体结构,晶格常数a = b = 0.320 nm,c = 0.513 nm;α = β = 90°,γ = 120° (α、β和γ分别为晶格常数a和b、a和c、b和c 3个轴之间的夹角).Mg2Sn析出相为fcc结构,a = b = c = 0.681 nm,α = β = γ = 90°.有实验观察到Mg2Sn与Mg基体之间的取向关系为//Mg(0001)取向(200℃左右)[12]和Mg(0001)//Mg2Sn(022)[4],但考虑到不同取向界面能不一样,本工作分别对Mg(0001)以及Mg2Sn 3个低指数面组成的界面进行第一性原理计算.经过收敛性测试,采用4层Mg2Sn和4层Mg构建了Mg(0001)/Mg2Sn(001)Mg端和Sn端界面、Mg(0001)/Mg2Sn(022)界面以及Mg(0001)/Mg2Sn(111) Mg端和Sn端界面,对其界面间距进行收敛性测试,并测量其界面能和晶格错配度,如表1所示. ...
Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set
2
1996
... 本工作所有计算均使用基于密度泛函理论的VASP[32]软件包进行.选用Perdew-Burke-Ernzerhof (PBE)的广义梯度近似(generalized-gradient-approximation,GGA)为电子交换关联泛函.经过收敛性测试,平面波截断能为400 eV.Monkhorst-Pack方案的k点最终设定为9 × 9 × 4和5 × 5 × 5用于Mg和Mg2Sn进行优化计算,4 × 4 × 1、5 × 3 × 1和3 × 2 × 1分别用于Mg(0001)/Mg2Sn(001)、Mg(0001)/Mg2Sn(022)和Mg(0001)/Mg2Sn(111)界面的计算.采用4 × 5 × 1、4 × 4 × 1、4 × 3 × 1和3 × 2 × 1分别对4层Mg2Sn(001)、4层Mg(0001)、4层Mg2Sn(022)以及4层Mg2Sn(111)表面进行计算.收敛参数设置:电子弛豫的收敛标准为10-5 eV,并且对于界面计算,结构优化收敛标准为结构内任何原子的作用力均小于0.1 eV/nm.考虑到表面原子间的相互作用,沿Z轴方向设置了长度为1 nm的真空层,使得2个相邻界面之间的相互作用可以忽略. ...
... 黏附功(Wad)反映了界面结构与2个表面结构之间的能量差[32].Wad可以反映界面结构的力学稳定性以及抵抗裂纹扩展的能力.稳定的界面结构对应于高的Wad.它被定义为单位面积上将界面分离成2个自由表面所需的能量[33]: ...
First-principle study on the influence of alloy elements on the stability of TiC/Mg interface
1
2020
... 黏附功(Wad)反映了界面结构与2个表面结构之间的能量差[32].Wad可以反映界面结构的力学稳定性以及抵抗裂纹扩展的能力.稳定的界面结构对应于高的Wad.它被定义为单位面积上将界面分离成2个自由表面所需的能量[33]: ...
合金元素对TiC/Mg界面稳定性影响的第一性原理研究
1
2020
... 黏附功(Wad)反映了界面结构与2个表面结构之间的能量差[32].Wad可以反映界面结构的力学稳定性以及抵抗裂纹扩展的能力.稳定的界面结构对应于高的Wad.它被定义为单位面积上将界面分离成2个自由表面所需的能量[33]: ...
Adhesion, bonding and mechanical properties of Mo doped diamond/Al (Cu) interfaces: A first principles study
1
2020
... 界面掺杂能(Ef)可以反映掺杂界面形成的难易程度,其值越小越容易掺杂[34]: ...
First-principle investigation on the interfacial structure evolution of the δ'/θ'/δ' composite precipitates in Al-Cu-Li alloys
1
2020
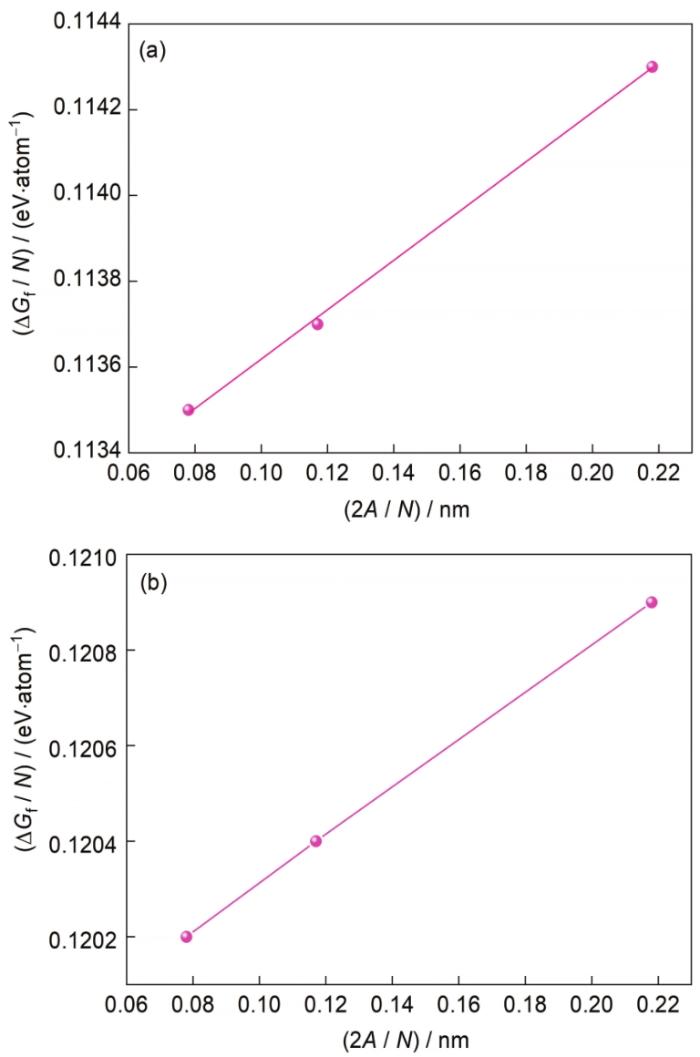
... 总的界面形成能包括界面能和弹性应变能.界面能是指Mg/Mg2Sn构建的界面体系分成独立晶体Mg和Mg2Sn所需的能量,而弹性应变能是指Mg基体和Mg的析出相Mg2Sn之间所需的应变能.考虑到界面周围最近原子层的影响,对Mg(0001)/Mg2Sn(022)界面进行扩胞,构建了原子个数分别为56、112和168的3个晶胞,计算总的界面形成能[35]: ...
A first-principles study on interfacial properties of Ni(001)/Ni3Nb(001)
1
2014
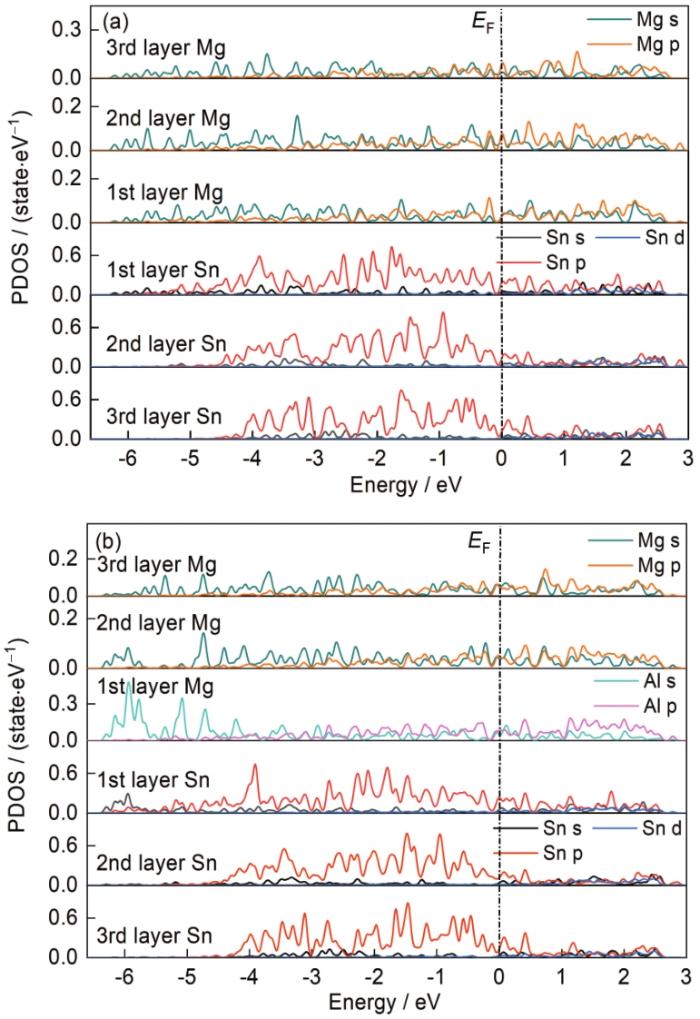
... 为了更深入地了解Mg(0001)/Mg2Sn(022)界面处掺杂Al原子的成键特性,绘制了掺杂Al元素前后Mg(0001)/Mg2Sn(022)界面的分波态密度(partial density of states,PDOS)曲线,如图4所示,图中黑色虚线代表Fermi能级(EF).分波态密度可以反映出界面处掺杂元素前后界面结构稳定性差异的物理本质[36].从图4a可以看出,第1~3层Mg和Sn在-4~0 eV处存在明显的重叠波峰,2者间存在较强的杂化共轭作用,界面处主要是Mg的s、p轨道与Sn的p轨道之间的相互作用,导致Mg、Sn原子之间形成较强的共价键.将图4a与b对比后发现,在界面第1层处添加Al元素后Mg、Sn、Al不仅在-4~0 eV处存在明显的重叠波峰,在-6.4~-4 eV处也出现了重叠波峰,并且Al的s轨道和Sn的p轨道存在明显的轨道杂化作用,使得Al掺杂界面的结合作用更强,结构更稳定. ...
Effects of transition element additions on the interfacial interaction and electronic structure of Al(111)/6H-SiC(0001) interface: A first-principles study
1
2021
... 基于对图5分析可以发现结构中存在广泛的共价键,但缺乏对这些共价键准确定量测量以及键强度和贡献的分析.晶体轨道Hamilton布居(crystal orbital Hamilton populations,COHP)可以定量分析晶体结构当中2个原子的结合强弱,而晶体轨道重叠布居积分值(integrated crystal orbital Hamilton populations,ICOHP)对应于Fermi能级积分能量值[37].图6a和b分别为hcp结构Mg和fcc结构Mg2Sn的投影晶体轨道Hamilton布居(projected crystal orbital Hamilton populations,pCOHP)[38]曲线,图6c~f为Mg(0001)/Mg2Sn(022)构建的界面以及在界面处掺杂Al的2种界面在界面处的Mg—Mg、Mg—Sn原子的pCOHP曲线.图中右侧的峰值(-pCOHP > 0)代表的是各个轨道成键贡献,左侧的峰值(-pCOHP < 0)代表的是各个轨道反键态的贡献.在这些原子相互作用中,比较图6c、d以及图6e、f,不难发现在界面处掺杂Al原子后,界面处Mg—Al之间以及Al—Sn之间都存在相互作用,主要是Mg3p、Al3p以及Sn3p、Al3p提供成键轨道.对于ICOHP的值排列如下:Mg3s—Sn5p > Mg3p—Al3p > Mg3s—Mg3p > Mg3p—Mg3p > Mg3p—Sn5s > Al3p—Sn5p,在界面处Al—Sn键强度占主导地位,Mg—Al键次之.通过将界面处原子之间相互作用分解为对不同轨道的贡献,对于不掺杂Al的界面,界面处Mg—Mg之间主要是3p轨道提供成键,Mg—Sn之间主要是Mg3p、Sn5s、Sn5p提供成键. ...
Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets
1
2011
... 基于对图5分析可以发现结构中存在广泛的共价键,但缺乏对这些共价键准确定量测量以及键强度和贡献的分析.晶体轨道Hamilton布居(crystal orbital Hamilton populations,COHP)可以定量分析晶体结构当中2个原子的结合强弱,而晶体轨道重叠布居积分值(integrated crystal orbital Hamilton populations,ICOHP)对应于Fermi能级积分能量值[37].图6a和b分别为hcp结构Mg和fcc结构Mg2Sn的投影晶体轨道Hamilton布居(projected crystal orbital Hamilton populations,pCOHP)[38]曲线,图6c~f为Mg(0001)/Mg2Sn(022)构建的界面以及在界面处掺杂Al的2种界面在界面处的Mg—Mg、Mg—Sn原子的pCOHP曲线.图中右侧的峰值(-pCOHP > 0)代表的是各个轨道成键贡献,左侧的峰值(-pCOHP < 0)代表的是各个轨道反键态的贡献.在这些原子相互作用中,比较图6c、d以及图6e、f,不难发现在界面处掺杂Al原子后,界面处Mg—Al之间以及Al—Sn之间都存在相互作用,主要是Mg3p、Al3p以及Sn3p、Al3p提供成键轨道.对于ICOHP的值排列如下:Mg3s—Sn5p > Mg3p—Al3p > Mg3s—Mg3p > Mg3p—Mg3p > Mg3p—Sn5s > Al3p—Sn5p,在界面处Al—Sn键强度占主导地位,Mg—Al键次之.通过将界面处原子之间相互作用分解为对不同轨道的贡献,对于不掺杂Al的界面,界面处Mg—Mg之间主要是3p轨道提供成键,Mg—Sn之间主要是Mg3p、Sn5s、Sn5p提供成键. ...








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}