
朱姜蕾, 王庆, 王海鹏
西北工业大学应用物理系 西安 710072
ZHU Jianglei, WANG Qing, WANG Haipeng
中图分类号: O469
文章编号: 0412-1961(2017)08-1018-07
通讯作者:
收稿日期: 2017-02-20
网络出版日期: 2017-08-20
版权声明: 2017 《金属学报》编辑部 《金属学报》编辑部
基金资助:
作者简介:
作者简介 朱姜蕾,女,1992年生,硕士生
展开
摘要
针对液态金属Cu在深过冷亚稳条件下的热物理性质和液态结构数据缺乏的问题,采用分子动力学方法结合修正嵌入原子势,研究了常规液态和亚稳液态金属Cu的热物理性质(熔点、密度、比热容和自扩散系数)和原子分布规律,体系温度范围为800~2400 K,最大过冷度达到556 K。通过构建晶体-液体-晶体结构,探索了金属Cu的熔化过程,获得最优的熔点计算温度为1341 K,与实验值误差1.11%。获得了宽广温度范围内液态金属Cu的密度随温度的变化规律,采用Mishin势函数计算的熔点处密度模拟值为7.86 g/cm3,与文献报道的实验结果的误差小于2%。液态金属Cu的焓在800~2400 K范围内随温度呈线性关系变化,即比热容几乎不随过冷度变化而变化,而自扩散系数则随温度呈指数关系变化。根据不同温度原子的位置变化,获得了相应的双体分布函数,发现液态体系始终处于短程有序、长程无序的状态,且原子短程有序度随温度升高而降低,短程有序结构仅保持在3~4个原子间距范围内,且随间距增大而展现出典型的无序特征。
关键词:
Abstract
Cu is commonly used in the field of electricity and electronics because of its high ductility, and electrical and thermal conductivity. The thermophysical properties and the atomic structure of liquid Cu, especially for undercooled state, are of practical significance in both application and fundamental researches. The major approaches to obtain thermophysical properties of undercooled metals are containerless techniques based on electrostatic levitation, electromagnetic levitation and ultrasonic levitation et al. However, the strong volatility of liquid Cu results in great difficulties to measure the thermophysical properties. Accordingly, computational prediction is becoming an expected method to obtain the thermophysical data of liquid Cu. The molecular dynamics (MD) simulation, in combination with a resonable potential model, has been extensively employed in studying the physical properties of several metals as a powerful approach. In this work, the atomic distribution and thermophysical properties including melting temperature, density, specific heat and self-diffusion coefficient of liquid Cu were studied by molecular dynamics simulation. Mishin's and Zhou's embedded-atom method potentials, and the modified embedded-atom method potential proposed by Baskes were used over the temperature range of 800~2400 K, reaching the maximum undercooling of 556 K. The simulated results are in good agreement with the reported experimental results. The crystal-liquid-crystal sandwich structure has been used to calculate the melting point. The melting point calculated by Baskes' potential model is 1341 K, just a difference of 1.11% from the experimental value. The density at the melting point calculated by Mishin's potential is 7.86 g/cm3, with a difference less than 2% compared with the reported data. It is found that the enthalpy of liquid Cu increases linearly with the increase of temperature. The specific heat is obtained to be 31.89 J/(molK) by Mishin's potential, which is constant in the corresponding temperature range. The self-diffusion coefficient is exponentially dependent on the temperature. The maximum error between the reported value and the present value of the self-diffusion coefficient calculated by Mishin's potential is only 4.93%. The pair distribution function was applied to investigate the atomic structure of liquid Cu, which suggests that the simulated system is still ordered in short range and disordered in long range for both normal liquid and undercooled state. It is found that the atomic ordered degree is weakened with the increase of temperature, and it is kept within 3~4 atom neighbor distance.
Keywords:
液态金属尤其是亚稳液态金属的热物理性质和原子分布规律是凝聚态物理、材料物理和流体物理领域的重要研究课题,与之相关的重要热物理性质主要包括密度、比热容、扩散系数、黏度和表面张力等。其中,密度是金属最基本的性质,而熔点附近密度变化规律对理解凝固过程十分关键;比热容在液态金属晶体形核和枝晶生长定量热力学和动力学计算中均不可或缺;扩散系数表征原子迁移过程和速度,由于需要跟踪同位素,液态金属自扩散的实验数据十分匮乏[1,2]。与热物理性质相比,液态金属原子分布信息则更为有限,这与其高温特性和原子的快速迁移及金属的不透明特征有关。双体分布函数被广泛应用于描述液态和非晶态的原子分布,是理论与实验对照的基本依据。因此探索液态金属的热物理性质和原子分布规律意义重大。
当液态金属冷却至熔点温度以下不发生凝固仍然保持液态的现象称为过冷状态,过冷液态金属在热力学上处于亚稳态[3]。当前研究液态金属深过冷较为有效的途径是无容器处理实验,如气动悬浮、电磁悬浮、静电悬浮和超声悬浮等[4~6]。Demmel等[7]采用气动悬浮装置实现了对Ni75Si25合金的深过冷;Hu等[8]采用静电悬浮技术使Zr91.2Si8.8合金最大过冷度达371 K;Perepezko和Imhoff [9]采用熔融玻璃净化法得到深过冷Au,并对液态Au的结晶控制进行研究。在热物理性质方面,实验测定仍然备受关注。Johnson等[10]采用静电悬浮技术研究了过冷Zr-Ni合金的密度和粘度;Ishikawa等[11]采用静电悬浮技术测定了稀土元素Gd的密度、表面张力和黏度;Wunderlich等[12]采用电磁悬浮技术测定了铁基合金的密度和表面张力等热物理性质。Cho等[13]、Brillo等[14]和Meyer[15]分别采用悬浮技术研究了液态Cu的密度和自扩散系数。尽管无容器实验能产生较大的过冷度,相关热物理性质研究已有报道,但实验测定热物理性质仍然存在很大困难。随着计算材料科学的发展,通过计算模拟方法获取深过冷液态金属的热物理性质令人期待。
分子动力学计算方法从20世纪60年代发展至今,已经成功应用于液态金属微观结构和热物理性质研究,且可提供模拟过程的原子坐标[16,17]。分子动力学计算方法可以精确控制冷却速率,实现实验难以达到的深过冷状态,具有广泛的适用性。但是,计算结果的可靠性和准确性一直是人们关注的重点,这主要依赖于势函数能否更为精确地描述原子间的相互作用关系。对于金属而言,简单立方和fcc结构的体系,其势函数比较可靠。金属Cu由于低电阻和高导热特性,普遍应用在电子电气等领域,而且Cu是fcc结构,非常适合采用分子动力学计算方法进行研究。Chen等[18]模拟了液态Cu熔点附近的自扩散系数;Wang和Wei[19]计算获得了温度范围900~2000 K时液态Cu的比热容和自扩散系数;Han等[20]计算获得了温度范围900~1900 K时液态Cu的比热容;Lü等[21]使用平衡和非平衡分子动力学方法对液态Cu输运性质进行了研究。以上研究从不同角度对液态Cu的性质进行了计算,得到了较为准确的模拟结果,不过这些研究主要集中在某些特定的热物理性质,对液态Cu仍缺少较为全面的描述,尤其是对液态Cu在原子尺度上的结构研究,多为定性的描述和表征;此外,实验中很难获得更宽温度范围,尤其是深过冷条件下的研究数据。
本工作使用3种不同的势函数,采用分子动力学计算方法,在较大温度范围内(800~2400 K),对液态Cu的密度、比热容和自扩散系数进行研究,分析热物理性质与温度的相互关系,进而计算双体分布函数,定量分析温度与其峰高的关系,探索液态短程结构的基本特征。
选用合适的势函数描述原子间相互作用对分子动力学模拟结果十分重要。嵌入原子法(embedded-atom method,EAM)由Daw等[22]在准原子近似理论[23]和有效介质理论[24]基础上提出,可较好描述金属原子间相互作用,但对非中心对称原子不适用。Baskes等[25~27]考虑了角度效应,提出了修正嵌入原子法(modified embedded-atom method,MEAM),不仅适用于中心对称的金属原子,也能较好地描述非中心对称的分子间相互作用。Mishin等[28]的EAM势使用多于传统方法的拟合参数,在有稳定原子构型情况下与实验吻合较好;Zhou等[29]的EAM势则使用初始设定的晶格常数与估算结构的内聚能算法;Baskes[25]的MEAM势的基本方程被改进并应用于包括密排六方结构在内的金属。本工作采用以上3种势函数对液态Cu的热物性进行分子动力学模拟,所有计算采用LAMMPS代码完成[30]。
密度、比热容和自扩散系数计算方法:建立包含4000个Cu原子的立方盒子,采用周期性边界条件,计算过程中使用恒温恒压(NPT)系综,压强为105 Pa,时间步长设置为1 fs,每隔50个时间步长对数据进行一次调整。为了得到稳定的液态结构,体系先在2500 K下驰豫200000步,在此基础上开始降温,每降低100 K进行弛豫200000步以达到平衡,统计该温度下的相关数据,直至降温到300 K。
采用晶体-液体-晶体结构法计算模拟熔点。为了产生晶体-液体-晶体结构共存的初始构型,建立包含8000个Cu原子的晶体-液体-晶体结构,上层和下层各包含2000个Cu原子,中层包含4000个Cu原子。使中层原子在2500 K下弛豫200000步,其余原子在1000 K下弛豫200000步,由此得到固液共存的构型。再使整个体系在不同设定温度下弛豫500000步。弛豫初期,体系为晶体-液体-晶体结构,随着弛豫时间增大,温度设定值在熔点以下时体系逐渐凝固,液态原子数减少,最终体系原子全部变为固态;温度设定值在熔点以上时与之相反,随着弛豫时间增大,固态原子逐渐熔化,液态原子数增多,最终全部变为液态。由于固液相转变伴随着能量的突变,统计在不同温度下系统充分弛豫后的内能,内能的突变点即为熔点。
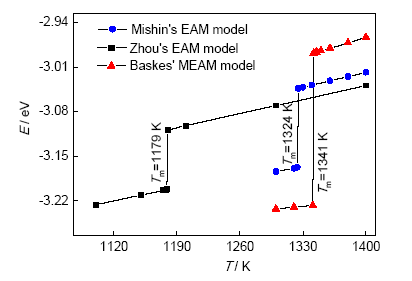
图1是采用3种势函数获得的体系平均每个原子的内能(E)随温度(T)的变化。可以看出,采用Mishin势函数得到的内能在1324~1325 K发生突变,由此确定E-T曲线突变的温度为(1324±1) K,即为熔点Tm。同理可得采用Zhou的势函数计算所得Tm为(1179±1) K;采用Baskes的势函数计算所得Tm为(1341±1) K。图2a和b分别表示了当体系温度在熔点以下和熔点以上时固液相变过程,初始状态为固液共存态,当温度低于熔点时,2个固液界面逐渐推进,最终合并,所有原子凝固成晶体;当温度高于熔点时,液相的Gibbs自由能较低,处于亚稳态的固相原子逐渐减少,最终体系停留在无序的液态。
图1 晶体-液体-晶体结构法计算Cu内能随温度的变化
Fig.1 Calculated curves of average internal energy vs temperature of Cu with a crystal-liquid-crystal structure using different potential models (T—temperature, Tm—melting temperature, E—energy)
图2 不同设定温度时体系相变过程
Fig.2 Simulated phase transition microstructure of Cu at T<Tm,Baskes (a) and T>Tm,Baskes (b) (Tm,Baskes—Tm using Baskes' MEAM model)
按照上述方法,采用Mishin、Zhou和Baskes 3种势函数计算的Tm分别为1324、1179和1341 K,与实验值1356 K[31]对比,误差分别为2.36%、13.05%和1.11%。其中采用Zhou的势函数计算出的熔点明显偏小,采用Baskes的势函数计算出的熔点误差最小。晶体-液体-晶体结构法的本质是在特定温度下比较液相与固相Gibbs自由能的大小,3种势函数,尤其是Mishin和Baskes的计算结果误差较小,表明它们能较为准确地描述液相和固相的自由能。然而计算结果均低于实际熔点,表明势函数所计算的液体自由能被低估,从而使得熔化更易发生。
采用3种不同势函数计算Cu的密度,结果如图3所示。本工作计算温度范围为800~2400 K,液态Cu最大过冷度为556 K,所得密度(ρ,g/cm3)与温度的关系为:
图3同时给出了本工作模拟值与实验值的对比[13,14,31,32]。Nasch和Steinemann[31]采用γ射线衰减法(gamma-attenuation technique,GAT)测得温度范围1356~1900 K时液态Cu的密度。Assael等[32]根据众多实验数据拟合,给出温度范围1356~2450 K的推荐值。用最小二乘法拟合获得数据,证实引文和本工作中采用EAM势函数所得液态Cu的密度值均随温度变化呈线性关系;MEAM势函数所得液态Cu的密度与温度呈二次关系。采用Mishin、Zhou和Baskes的势函数模拟所得熔点处密度分别为7.86、7.71和7.92 g/cm3,与Nasch等[31]的实验值8.02 g/cm3相比,误差分别为2.00%、3.87%和1.25%;与Cho等[13]的实验值7.90 g/cm3相比,误差分别为0.51%、2.41%和0.25%;与Assael等[32]在熔点处的参考值8.00 g/cm3相比,误差分别为1.73%、3.60%和0.98%。尽管不同研究者所测得的密度有所差异,但熔点附近的密度值都较为接近。显然,3种势函数在熔点处的密度模拟结果较为准确。2种EAM模型计算所得结果在1700 K以上时误差小于0.08 g/cm3,而在900~1600 K范围时,随温度降低误差变大,在900 K时误差为0.24 g/cm3。采用MEAM模型所得结果与采用2种EAM模型所得结果对比,温度在1700 K时3个模拟结果最接近;在1800~2400 K范围时MEAM模型的结果明显小于另外2种;在900~1500 K范围时MEAM模型的结果大于另外2种。采用Mishin势函数的计算结果与Cho等[13]、Brillo等[14]和Nasch等[31]的实验值以及Assael等[32]根据众多实验数据拟合的推荐值的最大误差仅为2.83%,比另外2种势函数的模拟值更接近实验值。不同势函数所得结果,在温度范围两端差异变大,而越靠近熔点差异越小。综上所述,在较大温度范围内,本模拟给出的液态金属Cu的密度具有较高精度(包含556 K的最大过冷度)。
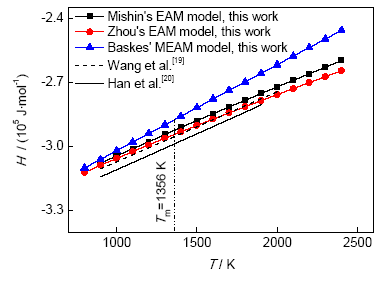
采用3种势函数计算所得焓与温度的变化关系如图4所示。液态Cu的焓随着温度的升高而增大,与温度呈线性关系,温度对液态Cu的比热容影响非常小,因此在一段温度范围内液态Cu比热容的值可视为常数。采用3种势函数所得焓(H,J/mol)与温度的线性拟合关系式分别为:
液态Cu比热容CPL的表达式为:
采用Mishin、Zhou和Baskes的势函数所得比热容的模拟值分别为31.89、29.57和40.16 J/(molK)。采用Mishin的势函数所得结果与Iida等[1]的实验值31.38 J/(molK) (温度范围1356~1600 K)最接近,误差仅为1.63%,和Han等[20]的模拟值33.66 J/(molK)(温度范围900~1900 K)误差为5.26%,和Wang等[19]的模拟值32.8 J/(molK) (温度范围900~2000 K)误差为2.77%;采用Zhou的势函数所得结果与Iida等[1]的实验值误差为5.77%,与Han等[20]的模拟值误差为12.15%,与Wang等[19]的模拟值误差为9.85%;而采用Baskes的势函数所得结果与Iida等[1]的实验值误差较大,为27.98%,与Han等[20]的模拟值误差为19.31%,与Wang等[19]的模拟值误差为22.44%。
自扩散系数(D,m2/s)由均方位移<r2(t)>(mean square displacement,MSD)计算获得:
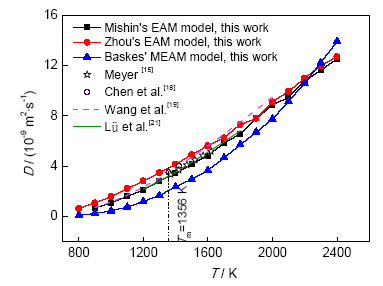
式中,ri(t)表示t时刻原子i的位置,原子位移的平方均值为MSD随时间的变化,可用来描述液态金属的扩散。当弛豫时间足够长,MSD近似于直线。根据Einstein扩散方程,由三维条件下MSD的斜率可计算体系的D。采用Arrhenius方程进行D-T拟合,结果如图5所示。
图5 液态Cu自扩散系数随温度的变化
Fig.5 Curves of self-diffusion coefficient (D) of liquid Cu vs T
采用3种势函数模拟值的指数拟合关系式分别为:
式中,R为气体常数。
Meyer[15]用准弹性中子散射法测定了1370~1620 K温度范围内液态Cu的D。Chen等[18]利用速度自相关函数(velocity-velocity correlation function,VCF)计算液态Cu熔点附近的D;Wang等[19]模拟了900~2000 K温度范围内的液态Cu的D,Lü等[21]模拟了1200~1800 K温度范围内液态Cu的D。如图5所示,将本研究数据与文献数据对比,在1370~1620 K温度范围内,Meyer[15]的实验值与采用Mishin的势函数模拟值误差仅为0.21%~4.93%,与采用Zhou的势函数模拟值误差范围为12.60%~15.42%,与采用Baskes的势函数模拟值误差范围为25.35%~42.64%;在熔点附近(1357和1423 K),Chen等[18]的模拟值与采用Mishin的势函数模拟值误差范围为11.00%~12.00%,与采用Zhou的势函数模拟值误差范围为6.25%~7.30%,与采用Baskes的势函数模拟值误差范围为43.50%~47.60%。在900~2000 K温度范围内,Wang等[19]的模拟值与采用Mishin的势函数模拟值误差范围为8.31%~17.28%,与采用Zhou的势函数模拟值误差范围为0.49%~27.04%,与采用Baskes的势函数模拟值误差范围为14.94%~75.54%。在1200~1800 K温度范围内,Lü等[21]的模拟值与采用Mishin的势函数所得模拟值误差范围为0.27%~6.65%,与采用Zhou的势函数模拟值误差范围为9.32%~20.79%,与采用Baskes的势函数模拟值误差范围为16.12%~53.59%。由图5可知,采用Mishin的势函数模拟值与实验值最接近;采用Zhou的势函数模拟值和Chen等[18]的模拟值最接近;在1500~2000 K温度范围内,采用Zhou的势函数模拟值和Wang等[19]的模拟值误差不超过4.09%。
除了Chen等[18]的研究结果,其它文献的自扩散系数均由MSD得到。由上述对比可知,对纯Cu这样的均一体系自扩散系数的计算,使用MSD和VCF 2种方法所得数据差异不大。计算VCF需要较长采样时间,且要对时间积分,因此对液态Cu来说,使用MSD计算自扩散系数效率更高。在800~2100 K温度范围内,采用Baskes的势函数模拟值始终小于2种EAM势函数的模拟值,2200 K时与采用2种EAM势函数的模拟值接近,2300~2400 K时明显超过采用2种EAM势函数的模拟值,由图5可知采用Mishin的势函数模拟值与实验值吻合较好。
双体分布函数g(r)可表示为:
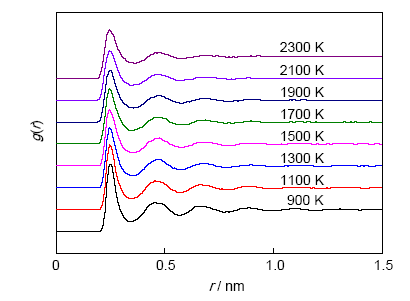
式中,V表示体积,ni(r, r+∆r)表示介于r到r+∆r距离之间球壳内的原子数目,N表示总原子数。相对采用Zhou和Baskes等的势函数,采用Mishin势函数计算所得的密度、比热容和自扩散系数都与实验值更接近,所以本工作使用由Mishin的势函数得到的g(r)来讨论液态Cu的原子分布。本工作模拟的g(r)既包括Cu的亚稳深过冷态,也包括常规液态,体系最低温度900 K,最大过冷度为456 K。
图6为900~2300 K温度范围内双体分布函数随温度的变化关系。可以看出,随r的增大,g(r)趋于1,这表明液态Cu在距离参考原子一定范围内才存在有序结构。在900 K时,g(r)的第三峰较为明显,同时可看到第四峰;而在1500 K以上时,第三峰和第四峰逐渐平缓;2100 K时,第四峰几乎消失。这表明液态Cu的短程有序结构仅保持在3~4个原子间距范围内。在图6中发现g(r)第一近邻处的值,随温度降低而增大,且第一峰越来越尖锐;第二近邻处g(r)的值,随温度的降低而增大。
图6 液态Cu在900~2300 K温度范围内双体分布函数
Fig.6 Pair distribution functions g(r) of liquid Cu at different temperatures (r—distance)
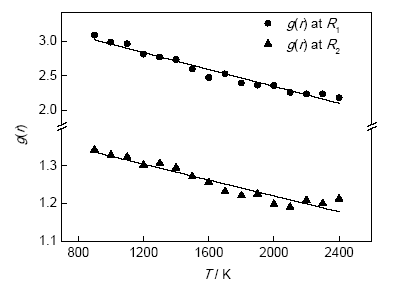
图7为液态Cu g(r)的第一近邻和第二近邻峰值随温度变化关系(R1表示第一近邻,R2表示第二近邻)。2400 K时,液态Cu在R1处的g(r)值为2.19,R2处的g(r)值为1.21;900 K时,液态Cu在R1处的g(r)值为3.09,R2处的g(r)值为1.34。2400 K时,R1处的值比900 K的值小29.12%,R2处的值比900 K的值小9.70%;1400 K时,液态Cu在R1处的g(r)值为2.73,显著低于最大过冷时的3.09,说明过冷态的Cu有序度比常规液态Cu的高;而R2峰值变化比R1小,说明液态结构主要变化出现在第一近邻处。
图7 液态Cu双体分布函数的第一近邻和第二近邻峰值随温度变化关系
Fig.7 Pair distribution functions g(r) as a function of T at the first and the second neighbor distance (R1 and R2) of liquid Cu
(1) 采用晶体-液体-晶体结构法研究固液界面变化,计算出Cu熔点的最优值为Tm,Baskes=(1341±1) K,与实验值误差为1.11%。
(2) 计算了液态Cu在800~2400 K、最大过冷度达556 K范围内的热物理性质。熔点处液态Cu的密度ρm,Baskes=7.92 g/cm3,与Cho等[13]的实验值误差为0.24%,2种EAM势函数所得密度与温度呈线性关系。发现液态Cu的焓随温度变化呈线性关系,通过体系的焓计算了液态Cu的比热容,其中CPL,Mishin=31.89 J/(molK)。液态Cu的自扩散系数随温度变化则呈指数关系。
(3) 采用Mishin的势函数模拟得到原子位置,进而获得液态Cu的g(r)随温度变化关系,结果表明液态Cu的短程有序结构距离为3~4个原子间距。
致谢 感谢空间材料科学与技术重点实验室主任魏炳波教授的支持,感谢博士生吕鹏在分子动力学模拟方面的帮助。
The authors have declared that no competing interests exist.

/
| 〈 |
|
〉 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}