


在过去的几十年里,随着全球能源消耗的快速增长和化石燃料带来的相关环境污染问题,人们致力于研究新型可再生能源和清洁能源载体。在所有替代品中,H2因其高能量密度、可持续性和无碳燃烧副产物[1~4]而成为最有希望的候选之一。目前,大部分H2是由化石燃料的蒸汽重整产生[5],其中的主要副产物CO2是不可避免的。因此,要使H2成为一种实用的无碳燃料,还需要无碳资源。电化学分解H2O为H2和O2是最理想的方法,在整个循环中,起始点和H2燃烧产物都是H2O。由于H2O的电解需要较大的正Gibbs能量,因此高效、低成本的析氢反应(HER)催化剂是实现氢经济的关键。Pt具有低的反应活化能、Tafel斜率和过电位,被认为是最有效的HER电催化剂[6~8]。但是,由于铂基催化剂的高成本和低储量,以降低Pt负载量为目标的设计和优化以及对相应机理的理解引起催化界的广泛关注[9~12]。
近年来,学者们主要利用吸附自由能和金属-氢键强度的计算来寻找性能优异的催化剂,它们都与催化剂的功函数、d带中心或d带形状等简单的物理参数有关[13]。科研人员[14~16]发现高效催化剂Pt、Au等是Z2拓扑金属,这表明拓扑能带结构与催化之间可能存在某种关系。近10年来,随着对拓扑材料中一些高效催化剂的研究,发现拓扑非平庸材料具有许多有趣的性质,如非平庸拓扑表面态、高载流子迁移率、手性Fermi子(Fermion)、超长的Fermi弧等。拓扑材料中受拓扑保护的表面态[17~19]和由线性能带交叉引起的高迁移率[20~23]在理论和实验上被证明可以有效调整表面吸附行为和异质催化反应中的电子转移,是高效催化剂的良好候选材料。虽然HER的化学反应发生在表面上,但表面本身强烈依赖于体带结构。于是,Xu等[24]提取了一个纯粹的本征物理参数,即投影Berry相位,它只依赖于体电子结构,且与HER的催化效率之间存在线性关系,可作为预测和设计HER催化剂的新型描述符。
1 理论方法和模型
本工作采用从头计算模拟软件包VASP[26,27]用于所有能量最小化计算,并使用经过充分优化的fcc Pt7Sb获得表面模型。所有DFT计算均采用广义梯度近似(GGA)下的Revised-Perdew-Burke-Ernzerhof (RPBE)交换关联泛函来完成[28]。采用高斯展宽(Gaussian smearing)方法[29]描述总能量,宽度设置为0.05 eV。计算过程中,分别将Pt_5d96s1、Sb_5s25p3和H_1s1轨道上的电子视为价电子。采用共轭梯度(CG)算法将离子弛豫到基态。收敛计算期间的平面波截断能、能量标准和力的阈值分别设置为400 eV、10-6 eV和0.1 eV/nm。对于Pt7Sb的体和平板模型,Brillouin区k点采样分别采用8 × 8 × 8和8 × 8 × 1网格的Monkhorst-Pack方案计算,所有平板模型的真空高度均为1.5 nm。选择具有不同终端的Pt7Sb平板模型,并考虑晶体结构的晶格对称性,如图1所示,A和B分别表示Pt-Sb混合终端和纯Pt终端的表面。所考虑的平板模型的表面能可通过下列公式计算[30~33],表面能(
式中,Eslab是5层的Pt终端或混合终端平板模型弛豫后的总能量,A是表面积,µPt和µSb分别是Pt7Sb体结构中Pt和Sb的化学势,
图1
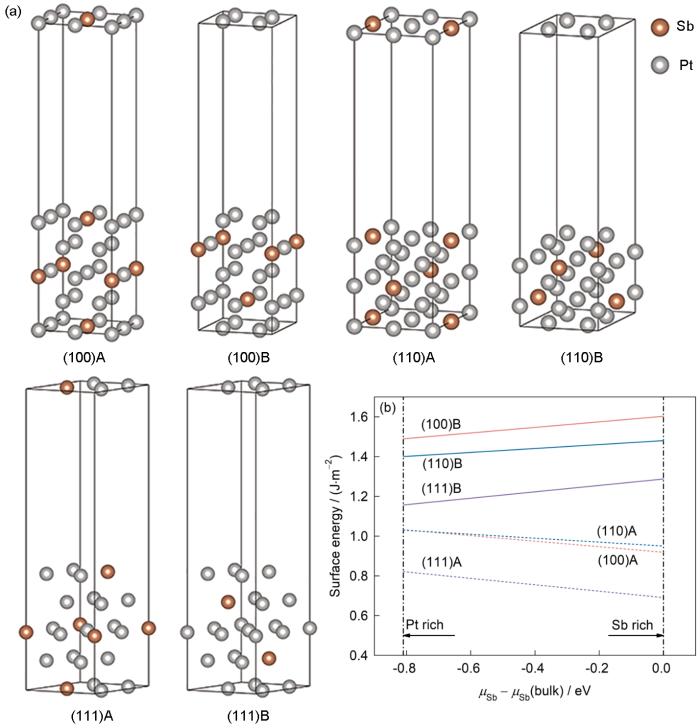
图1
具有不同晶格方向和终端的Pt7Sb平板模型和相应的表面能
Fig.1
Pt7Sb slab models with the different lattice orientations and terminations (a) and the corresponding surface energy variations with relative chemical potential (
Pt7Sb的形成能(ΔHf0)的定义如下:
当假设表面和体处于平衡状态时,体的化学势(
同时,利用式(
式中,
式中,k0是速率常数(设定为200 s-1·site-1),e是元电荷,kB是Boltzmann常数。
对于投影Berry相位的计算,方法参考文献[24]。
式中,
2 计算结果及讨论
2.1 晶体结构与表面能
图2
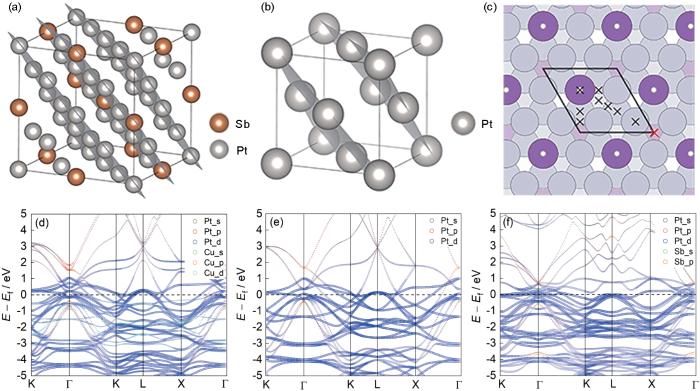
图2
Pt7Sb和Pt的晶体结构图,Pt7Sb(111)表面不同吸附位点示意图及Pt7Cu、Pt、Pt7Sb的能带结构
Fig.2
Crystal structure diagrams of Pt7Sb and Pt, schematic of different adsorption sites on Pt7Sb (111) surface, and the band structures of Pt7Cu, Pt, and Pt7Sb
(a, b) crystal structures of cubic Pt7Sb (a) and Pt (b)
(c) (111) surface of Pt7Sb with different surface adsorption sites labeled by crosses (The most stable site locates at the face-cubic center, as highlighted by the red cross)
(d-f) bulk band structures of Pt7Cu (d), Pt (e), and Pt7Sb (f), respectively (E is the energy, Ef is the Fermi level, and the horizontal coordinate represents the point of high symmetry in the inverse space)
为了进行对比分析,本工作还计算了与Pt7Sb结构完全相同的Pt7Cu。基于Pt7Sb与Pt7Cu原胞的晶体结构,计算了含有相同原子数量Pt的超胞的电子能带结构,并与Pt7Sb和Pt7Cu的电子能带结构进行了比较。如图2d~f所示,由于Pt7Cu和Pt7Sb可以被看作是在Pt超胞的基础上由Cu或Sb对Pt原子的有序替代掺杂而成,他们的能带结构必然会因为Cu或Sb轨道的引入在Pt的基础上发生变化。将图1f的Pt7Sb能带与图2e的Pt能带结构相比,Sb的s、p轨道均分布在远离Fermi能级的区域,且主要分布在深能处,由于没有d轨道的存在,使Pt元素整体能带相较于Fermi能级下移。虽然Pt7Sb和Pt具有相同的空间群,Sb原子对Pt原子的替代仍然会降低体系的对称性,使得Pt超胞中原来处于简并状态的能带在Pt7Sb中发生劈裂。这种劈裂使得Fermi能处的一些带宽变窄,尤其是Γ点附近,如图2f所示。与Pt7Sb类似,在Pt7Cu中,由于对称性的降低,能带发生了劈裂。由于Cu含有3d电子,与Pt7Sb相比,Pt7Cu的电子能带结构更接近与Pt,如图2d所示。
一般来说,表面能对材料的稳定性和化学性质有很大的影响,表面能越小,相应的表面就越容易形成。为了得到稳定的表面,沿着Pt7Sb的(100)、(110)和(111) 3个高对称方向构建平板模型,分别计算了其表面能。每个晶格方向主要有2种不同的情况,Pt-Sb混合终端(A)和纯Pt终端(B),详见图1。基于3种不同的晶格方向和2种不同的终端,按照
图1b给出了这6个表面的表面能与Sb的相对化学势的函数关系,其中正或负的斜率分别表示富Pt或富Sb的表面更稳定。它们都表现为随化学势变化的直线,并且上下排列,由此很容易确定Pt-Sb混合终端的(111)表面是Pt7Sb中最稳定的情况。作为参考,还计算了Pt7Cu的表面能。按照类似的方法,可以得出,对于Pt7Cu,在3个高对称方向中,纯Pt终端的(111)表面是最稳定的情况,如图3所示。对比计算Pt7Sb和Pt7Cu最稳定表面原子位置(即原子的Z坐标)的变化,发现Pt7Sb (111)B表面的2种Pt变化分别为-0.004689和-0.004691 nm,Sb变化为0.012883 nm,而Pt7Cu(111) A表面的2种Pt原子的变化分别为-0.001291 nm和0.002426 nm。同一表面原子位置的变化有正有负,这表明在所考虑的表面上发生了某些重构。并且值得注意的是,Cu和Pt的价电子均为s、d电子,而Sb原子的价电子为s、p电子,Pt-Sb混合表面的Pt_s轨道与Sb_p轨道之间可能存在一定的耦合,这不同于Pt7Cu的Pt_s轨道与次外层表面的Cu_d轨道之间的耦合,从而导致表面重构,间接影响表面能使其稳定表面产生差异。基于(111)取向的平板模型,分别分析Pt7Sb、Pt7Cu和Pt的相应表面HER催化性能。
图3
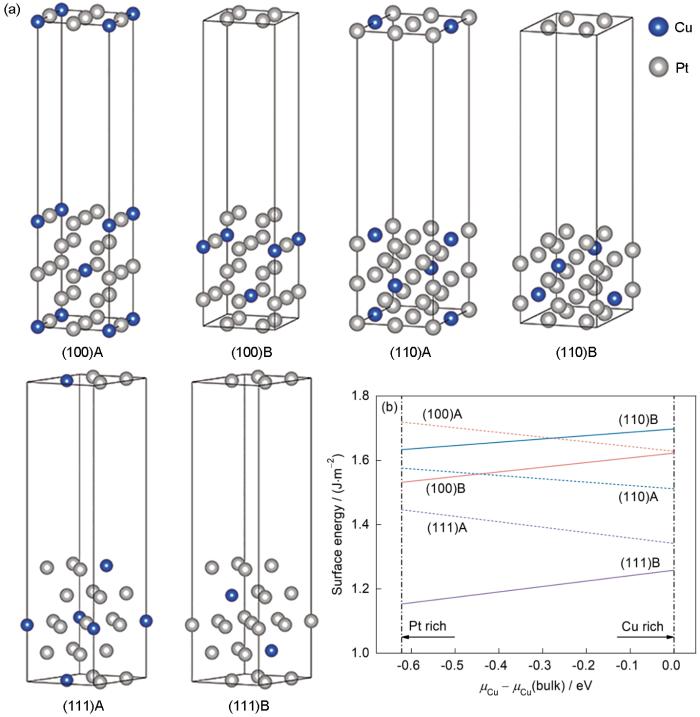
图3
具有不同晶格方向和终端的Pt7Cu平板模型和相应的表面能
Fig.3
Pt7Cu slab models with the different lattice orientations and terminations (a) and the corresponding surface energy variations with relative chemical potential (
2.2 HER性能
为了确定H原子在催化剂表面吸附时的相对稳定位置,利用
图4
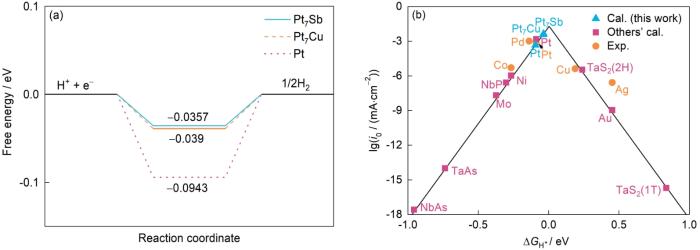
图4
Pt7Sb、Pt7Cu和Pt的台阶图及其与文献[20,37,42]对比所得火山图
Fig.4
Step diagrams of Pt7Sb, Pt7Cu, and Pt; and the volcano plot (exchange current density i0vs the free energy for hydrogen adsorption (
(a) calculated free energy diagram for hydrogen evolution reaction (HER) at a potential U = 0 V relative to the standard hydrogen electrode at pH = 0 (The free energy of H+ + e- is defined as the same as that of 1/2H2 at standard conditions)
(b) volcano plot for the HER of Pt7Sb and Pt7Cu in comparison with various pure metals (The experimental[37] and calculated[42] data of Pt, Pd, Co, Ag, and Cu), topological Weyl semimetals (The calculated data of NbP, NbAs, and TaAs[20]), and other candidates (the theoretical data of TaS2(2H) and TaS2(1T)[20])
2.3 电子结构
为了进一步理解Pt7Sb的良好HER性能,详细分析了其电子结构。以Pt7Sb为例,将Pt7Sb与元素形式的Sb和Pt体态密度(DOS)进行了比较。从图5a可以看出,Pt7Sb的DOS的形状接近于Pt,但与Sb非常不同。Pt7Sb可以被看作是在Pt超胞的基础上由Sb掺杂而成,并且Pt_5d轨道主导了Fermi能级附近的态。同样,Pt7Cu Fermi能级附近的态也是由Pt_5d轨道主导的,但Cu_3d轨道也做出了少量贡献,如图5b所示。这与前面关于电子能带结构的分析一致。基于这种理解,分析了表面有效区域(顶层与H相互作用的Pt原子)投影态密度(PDOS)的变化。图6a和b显示了Pt7Sb吸附H原子前后PDOS的变化。虽然Pt_5d轨道在H2吸附前后对Fermi能级附近的态的贡献占主导地位,但H2吸附后顶部Pt_5d态的PDOS明显下降。这种变化意味着电荷从Pt7Sb的表面转移到了H,同时形成一个弱化学键。图6c和d显示了Pt吸附H前后PDOS的变化,情况与Pt7Sb类似。对于Pt7Cu来说,Pt_5d轨道的PDOS在氢吸附后下降得更多,接近一个电子,如图6e和f所示,这是因为H在Pt7Cu表面的top位与其成键,仅有一个Pt原子与H发生相互作用,将电荷转移给H并形成了化学键。
图5

图5
Pt7Sb、Pt7Cu、Pt体结构电子态密度及差分电荷密度分析图
Fig.5
Bulk structure electronic densities of states and charge density difference analyses of Pt, Pt7Sb, and Pt7Cu (PDOS—projected electronic densities of states)
(a) PDOS for bulk Pt7Sb, fcc Pt, and Sb, respectively
(b) PDOS for bulk Pt7Cu, fcc Pt, and Cu, respectively
(c-e) top and side views of electron charge density differences for the hydrogen adsorption on Pt (c), Pt7Sb (d), and Pt7Cu (e) surfaces, respectively (Color code: Pt—grey; Sb—orange; H—red. The charge accumulation and depletion are depicted by the yellow and light blue regions, respectively. The isosurface levels are set to ± 0.002)
图6
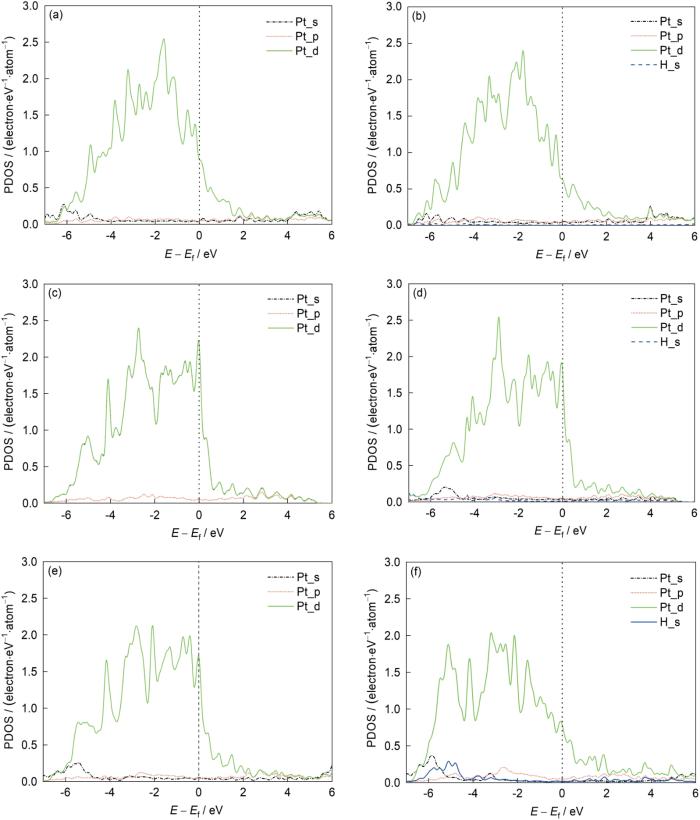
图6
Pt7Sb、Pt7Cu、Pt投影电子态密度图
Fig.6
PDOS for Pt7Sb (a, b), Pt (c, d), Pt7Cu (e, f) in the cases before (a, c, e) and after (b, d, f) hydrogen evolution
进一步,计算了Pt、Pt7Sb及Pt7Cu表面吸附H原子引起的实空间电荷密度差(Δρ)。Δρ通过
2.4 投影Berry相位
最近,有研究[24]提出了一个新的描述符——投影Berry相位。由于它只取决于本征的体能带结构,不受溶液pH值影响,具有相对稳定和普适性,并显示出与交换电流密度之间近乎线性的关系,投影Berry相位已成为了一个连接本征拓扑状态和催化性能的工具。为了解其对HER催化性能的本征贡献,本工作计算了Pt7Sb的投影Berry相位。由图7a投影Berry相位随能量的变化曲线可以看出,Pt7Sb Berry投影相位的峰值处于-0.5 eV附近,其绝对值可以达到0.09,与Pt的值比较接近。在Fermi能处,Pt7Sb Berry投影相位的绝对值可以达到0.065。在催化材料中,这是一个很大的数值,位于2个著名的高效HER催化剂Pd和Pt之间,如图7g所示,这也暗示Pt7Sb具有很高的催化效率。这也可以从Pt7Sb氢吸附Gibbs自由能的计算看出,如图4b所示。由于Pt和Pd的
图7

图7
Pt7Sb和Pt7Cu的投影Berry曲率和投影Berry相位
Fig.7
Projected Berry phases (γ) and Berry curvatures (Fxy ) projected of Pt7Sb and Pt7Cu
(a, d) the curves of the projected Berry phase (PBP) of Pt7Sb (a) and Pt7Cu (d) with energy
(b, c, e, f) electronic band structures (b, e) and local distributions of PBP (c, f) of Pt7Sb (b, c) and Pt7Cu (e, f)
(g) the theoretical results (red star) of PBP vs exchange current density of Pt7Sb in comparison with various pure metals (the experimental data (solid blue points) of Pt[43], Pd[44], Rh[45], Ir, Au, Cu[39], and Ag) and compounds (the theoretical data[24] of Pt7Cu (purple hollow dots)) with space group Fm
由于Pt7Sb与Pt7Cu具有相同的晶体结构,都相当于在Pt的基础上做其他元素的掺杂。相比于Pt7Sb,Pt7Cu的投影Berry相位更接近于元素Pt,因此Pt7Sb与Pt7Cu之间的对比有助于对Pt7Sb中的强投影Berry相位的理解。如图7d所示,Pt7Cu的投影Berry相位在Fermi能处有0.095左右,几乎等于Pt的值。与Pt7Sb不同之处在于投影Berry相位的峰值正好位于Fermi能级处,这与元素Pt比较类似。Pt7Cu的高效率催化性能也被表面氢吸附Gibbs自由能和直接的实验测量所验证[24]。在反应电流密度为10 mA/cm2的情况下,Pt7Cu的转换频率比商用20% Pt/C高出近5倍[24]。从2个材料的对比可以看出,不同元素掺杂的效果在细节上虽然不同,但是都能保持高的投影Berry曲率和小的氢吸附Gibbs自由能。
为了揭示Pt7Sb和Pt7Cu中强投影Berry相位的机制,本工作计算了这2种化合物沿着高对称方向的分布,并和相应的电子能带结构作了对比。如图7b和c所示,在高对称方向上,Pt7Sb投影Berry曲率局部分布有3个明显的峰值,分别处于L-Γ、Γ-K和X-Γ之间。结合电子结构可以看出,这3个极值对应的能带细节非常类似,都是不同能带间的线性反交叉,这种能带结构一个显著的特点是能够带来不同能带间的强耦合作用。这种效果在Pt7Cu中表现得更明显,尤其是Γ点附近。如图7e和f所示,由于自旋轨道耦合的作用,Γ点附近的能带简并被自旋轨道耦合作用打开局部带隙,导致了Fermi能附近能带的强烈耦合,并伴随着投影Berry曲率一个宽峰的出现。Pt7Sb和Pt7Cu中的强投影Berry相位主要是由这种自旋轨道耦合作用导致的退简并所导致。此外,这种能带结构经常伴随着拓扑电子态电子,因此Pt7Sb和Pt7Cu中的强投影Berry相位和其伴随的高效催化效率与其拓扑性存在着关联作用,属于材料的本征属性。
3 结论
从获得低Pt负载的良好催化剂的目标出发,找到了含Pt化合物Pt7Sb和Pt7Cu,并从理论上研究了其HER催化性能。计算结果显示,其
参考文献
Self-supported nanoporous cobalt phosphide nanowire arrays: An efficient 3D hydrogen-evolving cathode over the wide range of pH 0-14
[J].
In this Communication, we report the topotactic fabrication of self-supported nanoporous cobalt phosphide nanowire arrays on carbon cloth (CoP/CC) via low-temperature phosphidation of the corresponding Co(OH)F/CC precursor. The CoP/CC, as a robust integrated 3D hydrogen-evolving cathode, shows a low onset overpotential of 38 mV and a small Tafel slope of 51 mV dec(-1), and it maintains its catalytic activity for at least 80,00 s in acidic media. It needs overpotentials (η) of 67, 100, and 204 mV to attain current densities of 10, 20, and 100 mA cm(-2), respectively. Additionally, this electrode offers excellent catalytic performance and durability under neutral and basic conditions.
Nanostructured hydrotreating catalysts for electrochemical hydrogen evolution
[J].Progress in catalysis is driven by society's needs. The development of new electrocatalysts to make renewable and clean fuels from abundant and easily accessible resources is among the most challenging and demanding tasks for today's scientists and engineers. The electrochemical splitting of water into hydrogen and oxygen has been known for over 200 years, but in the last decade and motivated by the perspective of solar hydrogen production, new catalysts made of earth-abundant materials have emerged. Here we present an overview of recent developments in the non-noble metal catalysts for electrochemical hydrogen evolution reaction (HER). Emphasis is given to the nanostructuring of industrially relevant hydrotreating catalysts as potential HER electrocatalysts. The new syntheses and nanostructuring approaches might pave the way for future development of highly efficient catalysts for energy conversion.
An efficient and pH-universal ruthenium-based catalyst for the hydrogen evolution reaction
[J].The hydrogen evolution reaction (HER) is a crucial step in electrochemical water splitting and demands an efficient, durable and cheap catalyst if it is to succeed in real applications. For an energy-efficient HER, a catalyst must be able to trigger proton reduction with minimal overpotential and have fast kinetics. The most efficient catalysts in acidic media are platinum-based, as the strength of the Pt-H bond is associated with the fastest reaction rate for the HER. The use of platinum, however, raises issues linked to cost and stability in non-acidic media. Recently, non-precious-metal-based catalysts have been reported, but these are susceptible to acid corrosion and are typically much inferior to Pt-based catalysts, exhibiting higher overpotentials and lower stability. As a cheaper alternative to platinum, ruthenium possesses a similar bond strength with hydrogen (∼65 kcal mol), but has never been studied as a viable alternative for a HER catalyst. Here, we report a Ru-based catalyst for the HER that can operate both in acidic and alkaline media. Our catalyst is made of Ru nanoparticles dispersed within a nitrogenated holey two-dimensional carbon structure (Ru@CN). The Ru@CN electrocatalyst exhibits high turnover frequencies at 25 mV (0.67 H s in 0.5 M HSO solution; 0.75 H s in 1.0 M KOH solution) and small overpotentials at 10 mA cm (13.5 mV in 0.5 M HSO solution; 17.0 mV in 1.0 M KOH solution) as well as superior stability in both acidic and alkaline media. These performances are comparable to, or even better than, the Pt/C catalyst for the HER.
Nanoscale nickel oxide/nickel heterostructures for active hydrogen evolution electrocatalysis
[J].Active, stable and cost-effective electrocatalysts are a key to water splitting for hydrogen production through electrolysis or photoelectrochemistry. Here we report nanoscale nickel oxide/nickel heterostructures formed on carbon nanotube sidewalls as highly effective electrocatalysts for hydrogen evolution reaction with activity similar to platinum. Partially reduced nickel interfaced with nickel oxide results from thermal decomposition of nickel hydroxide precursors bonded to carbon nanotube sidewalls. The metal ion-carbon nanotube interactions impede complete reduction and Ostwald ripening of nickel species into the less hydrogen evolution reaction active pure nickel phase. A water electrolyzer that achieves similar to 20 mA cm(-2) at a voltage of 1.5V, and which may be operated by a single-cell alkaline battery, is fabricated using cheap, non-precious metal-based electrocatalysts.
A national vision of America's transition to a hydrogen economy: To 2030 and beyond
[A].
Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt interfaces
[J].Improving the sluggish kinetics for the electrochemical reduction of water to molecular hydrogen in alkaline environments is one key to reducing the high overpotentials and associated energy losses in water-alkali and chlor-alkali electrolyzers. We found that a controlled arrangement of nanometer-scale Ni(OH)(2) clusters on platinum electrode surfaces manifests a factor of 8 activity increase in catalyzing the hydrogen evolution reaction relative to state-of-the-art metal and metal-oxide catalysts. In a bifunctional effect, the edges of the Ni(OH)(2) clusters promoted the dissociation of water and the production of hydrogen intermediates that then adsorbed on the nearby Pt surfaces and recombined into molecular hydrogen. The generation of these hydrogen intermediates could be further enhanced via Li(+)-induced destabilization of the HO-H bond, resulting in a factor of 10 total increase in activity.
Nature of highly active electrocatalytic sites for the hydrogen evolution reaction at Pt electrodes in acidic media
[J].The hydrogen evolution reaction (HER) is one of the two processes in electrolytic water splitting. Known for more than two centuries, the HER still receives great attention in fundamental and applied science in view of its apparent simplicity (only two electrons are transferred), fast kinetics in acidic media, and promising technological applications in electrolyzers. However, the exact nature of active catalytic sites for this reaction is often uncertain, especially at nonuniform metal electrodes. Identification of such centers is important, as the HER will probably be central in future energy provision schemes, and it is simultaneously a convenient model reaction to study structure-composition-activity relations in catalysis. In this work, using simple coordination-activity considerations, we outline the location and geometric configuration of the active sites at various model Pt single-crystal electrodes. We show that when the coordination of such surface sites is optimized and their density at the surface is maximized, the experimental-specific HER activities are among the highest reported in the literature for pure platinum with a well-defined surface structure under similar conditions.
Cu3P-Ni2P hybrid hexagonal nanosheet arrays for efficient hydrogen evolution reaction in alkaline solution
[J].
Platinum nanoparticle during electrochemical hydrogen evolution: Adsorbate distribution, active reaction species, and size effect
[J].
Electrochemical activation of single-walled carbon nanotubes with pseudo-atomic-scale platinum for the hydrogen evolution reaction
[J].
Potential-cycling synthesis of single platinum atoms for efficient hydrogen evolution in neutral media
[J].Single-atom catalysts (SACs) have exhibited high activities for the hydrogen evolution reaction (HER) electrocatalysis in acidic or alkaline media, when they are used with binders on cathodes. However, to date, no SACs have been reported for the HER electrocatalysis in neutral media. We demonstrate a potential-cycling method to synthesize a catalyst comprising single Pt atoms on CoP-based nanotube arrays supported by a Ni foam, termed PtSA-NT-NF. This binder-free catalyst is centimeter-scale and scalable. It is directly used as HER cathodes, whose performances at low and high current densities in phosphate buffer solutions (pH 7.2) are comparable to and better than, respectively, those of commercial Pt/C. The Pt mass activity of PtSA-NT-NF is 4 times of that of Pt/C, and its electrocatalytic stability is also better than that of Pt/C. This work provides a large-scale production strategy for binder-free Pt SAC electrodes for efficient HER in neutral media.© 2017 The Authors. Published by Wiley-VCH Verlag GmbH & Co. KGaA.
Platinum single-atom and cluster catalysis of the hydrogen evolution reaction
[J].Platinum-based catalysts have been considered the most effective electrocatalysts for the hydrogen evolution reaction in water splitting. However, platinum utilization in these electrocatalysts is extremely low, as the active sites are only located on the surface of the catalyst particles. Downsizing catalyst nanoparticles to single atoms is highly desirable to maximize their efficiency by utilizing nearly all platinum atoms. Here we report on a practical synthesis method to produce isolated single platinum atoms and clusters using the atomic layer deposition technique. The single platinum atom catalysts are investigated for the hydrogen evolution reaction, where they exhibit significantly enhanced catalytic activity (up to 37 times) and high stability in comparison with the state-of-the-art commercial platinum/carbon catalysts. The X-ray absorption fine structure and density functional theory analyses indicate that the partially unoccupied density of states of the platinum atoms' 5d orbitals on the nitrogen-doped graphene are responsible for the excellent performance.
Theoretical surface science and catalysis-calculations and concepts
[J].
Topological states on the gold surface
[J].Yan, Binghai; Felser, Claudia Max Planck Inst Chem Phys Solids, D-01187 Dresden, Germany. Yan, Binghai Max Planck Inst Phys Komplexer Syst, D-01187 Dresden, Germany. Yan, Binghai ShanghaiTech Univ, Sch Phys Sci & Technol, Shanghai 200031, Peoples R China. Stadtmueller, Benjamin; Haag, Norman; Jakobs, Sebastian; Seidel, Johannes; Jungkenn, Dominik; Cinchetti, Mirko; Aeschlimann, Martin Univ Kaiserslautern, Dept Phys, D-67653 Kaiserslautern, Germany. Stadtmueller, Benjamin; Haag, Norman; Jakobs, Sebastian; Seidel, Johannes; Jungkenn, Dominik; Cinchetti, Mirko; Aeschlimann, Martin Univ Kaiserslautern, Res Ctr OPTIMAS, D-67653 Kaiserslautern, Germany. Mathias, Stefan Univ Gottingen, Inst Phys 1, D-37077 Gottingen, Germany.
Colloquium: Topological insulators
[J].
Topological insulators and superconductors
[J].
Fragility of surface states and robustness of topological order in Bi2Se3 against oxidation
[J].
Robust surface electronic properties of topological insulators: Bi2Te3 films grown by molecular beam epitaxy
[J].
Robustness of topological order and formation of quantum well states in topological insulators exposed to ambient environment
[J].The physical property investigation (like transport measurements) and ultimate application of the topological insulators usually involve surfaces that are exposed to ambient environment (1 atm and room temperature). One critical issue is how the topological surface state will behave under such ambient conditions. We report high resolution angle-resolved photoemission measurements to directly probe the surface state of the prototypical topological insulators, Bi(2)Se(3) and Bi(2)Te(3), upon exposing to various environments. We find that the topological order is robust even when the surface is exposed to air at room temperature. However, the surface state is strongly modified after such an exposure. Particularly, we have observed the formation of two-dimensional quantum well states near the exposed surface of the topological insulators. These findings provide key information in understanding the surface properties of the topological insulators under ambient environment and in engineering the topological surface state for applications.
Weyl semimetals as hydrogen evolution catalysts
[J].
Topological quantum catalyst: Dirac nodal line states and a potential electrocatalyst of hydrogen evolution in the TiSi family
[J].
Dirac nodal arc semimetal PtSn4: An ideal platform for understanding surface properties and catalysis for hydrogen evolution
[J].
Extremely large magnetoresistance and ultrahigh mobility in the topological Weyl semimetal candidate NbP
[J].Shekhar, Chandra; Nayak, Ajaya K.; Sun, Yan; Schmidt, Marcus; Nicklas, Michael; Schnelle, Walter; Borrmann, Horst; Grin, Yuri; Felser, Claudia; Yan, Binghai Max Planck Inst Chem Phys Solids, D-01187 Dresden, Germany. Nayak, Ajaya K. Max Planck Inst Microstruct Phys, D-06120 Halle, Germany. Leermakers, Inge; Zeitler, Uli Radboud Univ Nijmegen, HFML EMFL, NL-6525 ED Nijmegen, Netherlands. Skourski, Yurii; Wosnitza, Jochen Helmholtz Zentrum Dresden Rossendorf, Dresden High Magnet Field Lab HLD EMFL, D-01328 Dresden, Germany. Liu, Zhongkai Diamond Light Source, Harwell OX11 0QX, Berks, England. Chen, Yulin Univ Oxford, Dept Phys, Oxford OX1 3PU, England. Yan, Binghai Max Planck Inst Phys Komplexer Syst, D-01187 Dresden, Germany.
Descriptor for hydrogen evolution catalysts based on the bulk band structure effect
[J].The vital role of electrocatalysts in determining the efficiency of renewable energy conversion inspired the uncovering of the relation between the catalytic efficiency and electronic structure, in which the volcano-type plot based on adsorption energies and d-band model has achieved great success. At the same time, catalysts with nontrivial topological electronic structures have received considerable attention because of their robust topological surface states and high-mobility electrons, which favor the electrons transfer processes in the heterogeneous catalysis reactions. Under the guidance of this theory, excellent catalysts were reported among topological materials. Inspired by the current development of catalyst and topological materials, we tried to extract a pure intrinsic physical parameter, projected Berry phase (PBP), that only depends on the bulk electronic structure. Applying this parameter to the well-known nonmagnetic transition-metal electrocatalysts, we found a linear relationship between PBP and catalytic efficiency of hydrogen evolution reaction (HER) after considering the symmetry constraint. This can be used as a descriptor for the prediction and designing of promising catalysts for HER, which is realized experimentally in PtCu nanostructures. This work illustrates the importance of the pure bulk band structure effect on electrochemical activities and implies an effective way to understand the mechanism of HER catalysts.Copyright © 2020 American Chemical Society.
Co oxidation facilitated by robust surface states on Au-covered topological insulators
[J].
Ab-initio simulations of materials using VASP: Density-functional theory and beyond
[J].
A search for two types of transverse excitations in liquid polyvalent metals at ambient pressure: An ab initio molecular dynamics study of collective excitations in liquid Al, Tl, and Ni
[J].
Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals
[J].
High-precision sampling for Brillouin-zone integration in metals
[J].
Consistent methodology for calculating surface and interface energies
[J].
Stoichiometry and adhesion of Nb/Al2O3
[J].
Adhesion, stability, and bonding at metal/metal-carbide interfaces: Al/WC
[J].
Abinitio calculations on the Al2O3(0001) surface
[J].
Mechanism of hydrogen evolution reaction on 1T-MoS2 from first principles
[J].
Widely available active sites on Ni2P for electrochemical hydrogen evolution—Insights from first principles calculations
[J].
Computational screening of electrocatalytic materials for hydrogen evolution: Platinum monolayer on transitional metals
[J].
Trends in the exchange current for hydrogen evolution
[J].
Les systèmes binaires Pb Sb et Pt Sb
[J].
Work function, electronegativity, and electrochemical behaviour of metals: III. Electrolytic hydrogen evolution in acid solutions
[J].
Sur une manière de décomposer l'eau en air inflammable et en air vital
[J].
Hydrogenation and dehydrogenation by catalysis
[J].
Computational high-throughput screening of electrocatalytic materials for hydrogen evolution
[J].The pace of materials discovery for heterogeneous catalysts and electrocatalysts could, in principle, be accelerated by the development of efficient computational screening methods. This would require an integrated approach, where the catalytic activity and stability of new materials are evaluated and where predictions are benchmarked by careful synthesis and experimental tests. In this contribution, we present a density functional theory-based, high-throughput screening scheme that successfully uses these strategies to identify a new electrocatalyst for the hydrogen evolution reaction (HER). The activity of over 700 binary surface alloys is evaluated theoretically; the stability of each alloy in electrochemical environments is also estimated. BiPt is found to have a predicted activity comparable to, or even better than, pure Pt, the archetypical HER catalyst. This alloy is synthesized and tested experimentally and shows improved HER performance compared with pure Pt, in agreement with the computational screening results.
The mechanism of the hydrogen evolution reaction on platinum, silver and tungsten surfaces in acid solutions
[J].
Hydrogen evolution reaction on copper, gold, molybdenum, palladium, rhodium, and iron: Mechanism and measurement technique under high purity conditions
[J].
Elucidation of the mechanism of electrolytic hydrogen evolution by the use of H-T separation factors
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}