
中图分类号: TG111.1,TG111.2
文章编号: 0412-1961(2019)02-0223-06
通讯作者:
收稿日期: 2018-08-20
网络出版日期: 2019-01-31
版权声明: 2019 《金属学报》编辑部 《金属学报》编辑部
基金资助:
作者简介:
作者简介 陈丽群,女,1964年生,教授,博士
展开
摘要
利用DMol和离散变分法,研究了Ru在NiAl [100](010)刃型位错中择优占位和合金化效应。杂质偏聚能的计算结果表明,Ru会优先占据Al心位错芯中的Al格位。原子间相互作用能、电荷密度和态密度的分析表明,杂质原子和相邻基体原子之间形成了较强的化学键,使Ru原子与位错芯区近邻基体原子间因强相互作用形成一个整体。此外,在掺杂体系中,穿过滑移面的基体原子间相互作用减弱,而沿滑移方向基体原子间的相互作用加强。这样的成键特性有利于位错线沿滑移面的移动形成扭折,扭折的成核及迁移促使位错运动,从而改善NiAl合金的韧性。
关键词:
Abstract
NiAl intermetallics have potential application in the aerospace industry as a new high temperature structure material due to its high melting temperature, good thermal conductivity, low density, and good oxidation resistance. However, possible technological applications of NiAl are limited by its poor ductility at low temperatures and brittle grain boundary fracture at elevated temperature. Different methods have been dedicated to manage the brittle behavior of NiAl. Micro-alloying is a effective method. Dislocation is a complicated and widely existing crystal defect. The interaction between dislocation and impurity can greatly influence the mechanical properties of materials. However, the mechanism of interaction between the dislocation and alloying element is not clear. In the work, using the DMol and the discrete variational method within the framework of density functional theory, the site preference and alloying effect of Ru in the [100](010) edge dislocation core (DC) of NiAl are studied. The results of the impurity formation energy show that Ru exhibits a strong Al site preference. The analyses of the interatomic energy, the charge distribution and the partial density of states show that the strong bonding states are formed between the impurity atom and neighboring host atoms. Meanwhile, the bonds keep the atoms in the DC as a whole, which will benefit formation of kink. In addition, in the doped DC system, the interactions between the pair of atoms across the slip plane are weaker, while along the slip direction the interactions are stronger than those in the clean DC system. This bond characters may be in favor of the motion of [100](010) edge dislocation, which will improve the ductility of NiAl.
Keywords:
B2超晶格结构的金属间化合物NiAl具有诸多优异特性,如高达1638 ℃的高熔点,5.9 g/cm3相对低的密度,76 W/(mK)的高热导率(热导系数)[1],及在高达1400 ℃时还具有极好的抗氧化性能[2],因此,有可能成为高温轻型结构材料而替代镍基超合金应用在航空工业及耐高温涂层等领域。然而,由于受到NiAl本身较低的室温断裂韧性及高温强度的限制[1],导致其难以实际应用。在过去的20多年间,科研工作者[3,4,5,6,7]为了改善NiAl的性质,在加工处理及设计方法等方面做了大量有意义的工作,其中包括通过晶粒细化、单晶、微观和宏观合金化以及混合的第二相强化、燃烧合成及热等静压等方法来克服上述缺点。研究发现,微合金化是改善NiAl室温韧性和高温强度的有效方法之一。Zhang等[8]研究结果表明,随着Mo含量的增加,Mo纤维间距减少,体积分数增加,因而改善了NiAl的断裂韧性,阻止了裂纹的生长。Adharapurapu等[9]研究发现,Hf、Pd及Pt添加物的增加能够改善NiAl抗氧化性能,并能大大延长氧化寿命。Zr的添加会影响NiAl的微观结构及压缩塑性[10]。
Levit等[11]研究发现,不仅杂质含量和热处理会影响NiAl的力学性能,结构缺陷同样对其性能产生极大的影响。Han等[12]研究了定向凝固(directionally solidified, DS) NiAl-Mo(Hf) 的微观结构、高应变速率、超塑性及拉伸蠕变行为,发现这种合金呈现出高应变速率的超塑性形变行为,在1373 K最大的拉伸延伸率达104.2%,并且应变速率为1.04×10-2 s-1;同时也发现这种超塑性形变机制源自于位错滑移引起的应变硬化和动态恢复以及动态再结晶引起的应变软化之间的平衡。Wang 等[13]使用扫描电镜(SEM)、透射电镜(TEM)等方法,在不同的提拉速率下,对DS NiAl-Cr(Mo)-(Hf, Dy)合金的微观结构、室温断裂韧性及高温强度进行了研究,结果发现室温断裂韧性很高,这种情况的出现主要包括外部韧化机制和内部韧化机制,而内部韧化机制主要是NiAl相中位错的产生所起的作用。可见,位错的形成及滑移对材料的性能有显著的影响,同时位错也会与掺杂元素产生交互作用,进一步影响其性能。一些研究[14,15,16]发现,在位错芯区由于位错与掺杂元素间的相互作用而改善了NiAl的强度。虽然科研工作者为改善NiAl性能做了大量的工作,遗憾的是NiAl化合物在航空工业方面的应用仍未实现[17],以致在21世纪初有关NiAl 材料发展的科学报道有所放缓,但在最近的几年间,由于在航空工业对先进材料解决发展需要的刺激下,特别是一些性能优异的合金元素,如Ru[18]、Re[19]等的加入大大改善了NiAl的力学性能,目前,以Ru添加为标志的第四、五代镍基单晶高温合金已成为国际上该领域的研究热点。Wang等[20]研究发现,NiAl金属间化合物中其刃型位错类型绝大多数是Burgers矢量为b=[100]。因此,本工作选择NiAl[100](010)刃型位错模型,利用第一性原理研究Ru对NiAl[100](010)刃型位错电子结构的影响。首先利用分子动力学方法建立[100](010) 刃型位错模型;然后用DMol方法[21,22]弛豫2个掺Ru模型,根据杂质形成能确定合理稳定的计算模型;最后使用离散变分方法[23,24]计算原子间相互作用能、电荷密度及态密度,通过分析获得Ru与位错之间的相互作用对NiAl力学性能的影响机制。
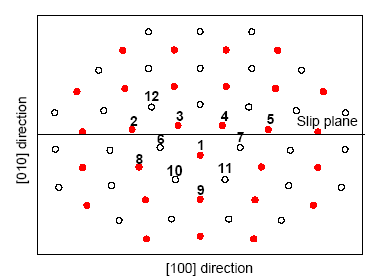
鉴于位错的一些重要性质可成功地利用弹性理论来描述,通过解能够代表刃型位错形变性质的双调谐方程获得刃型位错的初始原子构型[25]。为了获得位错的平衡原子构型,选择Voter-Chen-type[26]原子间势函数,利用分子动力学方法对位错初始原子构型进行弛豫,得到具有mm2对称性的[100](010)刃型位错芯平衡结构,如图1所示,其中实心圆和空心圆分别表示原子沿位错线[001]方向顺序堆垛的2个相邻平面,分别称为A平面和B平面。为了研究杂质原子Ru对NiAl合金力学性能的影响,在位错芯中引入单杂质原子,考虑结构对称性,Ru原子取代图1中标注序号为1的原子。杂质引入会引起结构局域畸变,因此应用DMol方法对掺杂模型进行结构优化,选取一个沿位错线方向即[001]方向包含7个原子面、总原子数为209的位错芯团簇,其中包含3个A平面和4个B平面。因为A、B平面上的原子既可以是Ni原子,也可以是Al原子,因此可以构成2种位错芯模型NiAlNiAlNiAlNi和AlNiAlNiAlNiAl,将这2种模型分别称之为Al心位错芯和Ni心位错芯。在优化过程中,只对序号为1~11的原子及等价原子进行弛豫,其它原子固定,弛豫的结果获得仍具有mm2对称性的掺杂模型。
图1 NiAl中[100](010)刃型位错芯的原子模型示意图(分别用实心圆和空心圆表示沿[001]方向2个相邻平面上的原子,称为平面A和平面B)
Fig.1 Atomic model of the [100](010) edge dislocation core in NiAl (Atoms denoted by solid circles and open circles construct two adjacent planes (plane A and plane B, respectively) in the stacking sequence along [001])
同时对上述2个模型分别计算了掺杂位错体系和未掺杂位错体系(称为纯位错体系)的结合能(Eb),其定义为
原子间相互作用能能够定量地计算出相邻2原子间的相互作用强度,能够说明2个原子间的成键情况。一般来说,原子间相互作用能是负值,其绝对值越大,则原子间相互作用越强。原子l和m间的相互作用能Elm的定义[29,30]为:
其中,i为分子轨道指标,Ni是分子轨道ψi的电子占据数;α和β分别为原子l和m的原子轨道;a*iαl=<ϕαl|ψi>,aiβl=<ϕβl|ψi>,是分子轨道展开成原子轨道ϕαl线性组合时的展开系数;Hαlβm=<ϕαl|
表1 掺杂位错体系和纯位错体系中被选定原子对的原子间相互作用能
Table 1 Interatomic energies for selected atomic pairs in the dislocation core (DC) with and without impurity
| Atomic-pair | E / eV | E′ / eV | ΔERu / eV |
|---|---|---|---|
| Atom1-Ni6 | -1.35 | -1.45 | -0.10 |
| Atom1-Ni10 | -1.03 | -1.32 | -0.29 |
| Atom1-Al3 | -0.71 | -1.44 | -0.73 |
| Al2-Al8 | -0.25 | -0.14 | 0.11 |
| Al2-Al3 | -1.03 | -0.82 | 0.21 |
| Al3-Al4 | -1.50 | -1.78 | -0.28 |
| Ni10-Ni11 | -0.93 | -1.10 | -0.17 |
从表1的数据分析得到,相比于纯位错体系,在掺杂位错体系中杂质Ru及其相邻的基体原子Atom1-Ni6、Atom1-Ni10及Atom1-Al3间的相互作用能绝对值明显增加,其中,Ru与最近邻基体原子Al3 (及与Al3等价的Al4原子)之间的相互作用能是纯位错体系中对应原子对之间的相互作用能的2倍。这些强相互作用验证了之前获得的掺杂体系的结合能远低于纯位错体系的结合能,使掺杂体系更稳定。另外,杂质原子与最近邻基体原子间这种强相互作用也表明,位错芯中这些原子可看作一个整体,在位错滑移时作整体迁移。
再看穿过滑移面的原子对,如Al2-Al8,在掺杂位错体系中的相互作用能要小于纯位错体系;而沿着滑移平面方向,掺杂位错体系中的原子对Al3-Al4和Ni10-Ni11之间的相互作用比纯位错体系更强,而Al2-Al3之间的相互作用较弱,这是因为Al3-Al4之间的强相互作用使得Al2-Al3之间的距离增大所致。这种沿滑移面方向原子间相互作用加强,而穿过滑移面原子间相互作用减弱的特点,使得位错沿滑移面的滑移更加容易。
由于掺杂体系中位错芯原子之间的相互作用有上述特点,这使得包含杂质原子的位错线段作为整体容易沿滑移面运动,以致在杂质处形成扭折,而位错则恰好通过原子尺度的扭折形成和迁移平滑地运动。NiAl合金低温塑性差的原因之一是滑移系太少,而通过原子间相互作用力的分析得到杂质Ru的掺入使得位错芯体系中[100](010)刃型位错的运动比在纯位错体系中更容易,Ru激活了NiAl中更多滑移系,这将会有利于NiAl合金延展性的改善,该结果与文献[18]的结果一致。
为了更直观形象地描述原子间相互作用,计算了位错芯区原子的电荷密度,电荷密度非常直观地给出了体系的电荷分布情况。电荷密度差分可通过从掺杂位错体系的电荷密度中减去纯位错体系的电荷密度来得到。为了更清晰地认识杂质引起电荷的重新分布,再减去自由的杂质原子及基体Ni原子和Al原子的电荷密度,得到二次密度差分。包含了杂质原子Ru的(001)原子面差分电荷密度分布如图2所示。
图2 掺杂位错体系包含杂质原子Ru的(001)原子面差分电荷密度分布 (等高线间隔为0.002e/(a.u)3,失去电荷以及得到电荷分别由虚线以及实线来表示)
Fig.2 Charge density difference of the (001) plane including impurity in the doped dislocation system (The contour spacing is 0.002e/(a.u)3. Solid and dashed lines mean a gain and loss of charge, respectively)
从图2中可以看出,除Ru原子周围有大量电荷分布外,其它原子几乎没有电荷分布,因为二次差分将这些原子的电荷密度减掉了,也就是杂质原子Ru对远离它的原子电荷分布几乎没影响,这表明了杂质影响的局限性。另外,因Ru的价电子数比Al多,所以Ru替换Al后在其周围有大量电荷聚集,特别在Ru原子与Al3和Al4原子间有许多电荷分布,这使得Ru原子与Al3和Al4原子之间的相互作用相较于纯位错体系中相应原子的原子间相互作用大大加强,这意味着杂质原子Ru和相邻的Al3(Al4)原子之间形成了很强的化学键,这些原子因此而形成一个整体,有利于包含杂质原子Ru位错线段的滑移。这个结果与原子间相互作用是一致的。
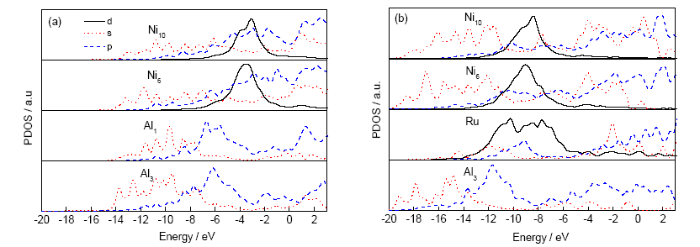
为了更详细地分析基体原子和杂质原子之间的相互作用,计算了位错芯区杂质原子Ru和杂质原子的最近邻Ni6、第二近邻Ni10及Al3基体原子的分波态密度(partial density of state,PDOS)。在离散变分方法中应用Lorentz展宽方法[29],可得原子轨道的分波态密度。图3a和b分别为纯位错体系中原子的分波态密度和掺Ru位错体系对应原子的分波态密度。
对比图3a和b,可以看出,掺杂体系中各原子分波态密度较纯位错体系中对应原子的分波态密度都向深能级移动了5~8 eV,特别是原子Ni6和Ni10的d轨道和原子Al3的s轨道移动的幅度更大,这意味着掺杂体系中这些原子的自由电子从价带中转移到更深的能级,并与杂质原子Ru及基体原子的价电子形成了较强的化学键,使掺杂体系更加稳定,杂质原子与基体原子间的相互作用加强。
此外,从图3a还可以看出,在纯位错体系中,位错芯中心原子Al1的s轨道在-12~-7 eV和p轨道在-7~-3 eV与Ni6、Ni10、Al3的s轨道和p轨道重叠,而Al1的s、p轨道与Ni6、Ni10的d轨道几乎没有重叠。所以,在纯位错体系中,中心原子Al1与近邻Ni6、Ni10、Al3原子的相互作用主要是来自s、p轨道的杂化。由图3b可看出,在掺杂体系中,Ru原子的d轨道在-12~-7 eV与Ni6、Ni10原子的d轨道有重叠,与Al3的p轨道重叠;Ru原子的s轨道在-14~-10 eV和-3~-1 eV与Ni6、Ni10原子的s轨道及Al3的p轨道有重叠;Ru原子的p轨道在-12~-8 eV和-4~3 eV与Ni6、Ni10、Al3原子的p轨道重叠。可见杂质Ru原子与近邻基体Ni6、Ni10原子之间的相互作用主要缘于它们之间的spd轨道杂化,而Ru与Al3原子之间的相互作用则主要来自于Ru的spd轨道与Al3的p轨道之间的杂化,因d轨道的价电子数较多,所以Ru原子与Ni6、Ni10原子间的相互作用强于Ru与Al3原子间的相互作用。同时杂质原子Ru与基体原子Ni6、Ni10、Al3之间的相互作用也比纯位错体系中中心Al1原子与其近邻Ni6、Ni10、Al3的相互作用更强。这些结果与电荷密度、原子间相互作用能的计算结果一致。
图3 纯位错体系和Ru掺杂的掺杂位错体系中杂质原子和其近邻原子的分波态密度(PDOS)曲线 (Fermi能级平移到零,图中阿拉伯数字对应于
Fig.3 Partial densities of state (PDOS) curves for impurities and neighboring host atoms in the clean DC (a) and the Ru-doped DC (b) (The Fermi level is shifted to zero. The Arabic numbers correspond to those in
(1) 通过计算杂质形成能发现Ru降低了体系的总能,使掺杂体系比纯位错体系更稳定,位错芯对杂质Ru有捕获效应。比较2个掺杂模型的杂质形成能,发现Ru原子优先占据Al心位错芯中的Al格位。
(2) 分析Al心位错芯模型的原子间相互作用能和电荷密度表明:与纯位错体系相比,在掺杂体系中杂质原子与最近邻原子间相互作用加强,使位错芯形成一个整体;另外,沿滑移方向基体原子间相互作用也加强,但穿过滑移面的原子对相互作用减弱,这样的成键特性有利于包含杂质原子位错线段优先沿滑移面移动形成扭折,扭折的成核及迁移促使位错运动,增加了NiAl合金的滑移系,从而改善NiAl合金的韧性。
(3) 态密度的计算进一步说明:由于杂质原子Ru的spd轨道与Ni原子spd轨道和Al原子的p轨道杂化,导致杂质原子与近邻基体原子间的相互作用增强;此外,从态密度分布还发现:与纯位错体系比较,掺杂位错体系中近邻原子的价电子都向更深能级移动且参与成键,使掺杂体系更稳定。总之,杂质原子Ru引入到位错芯中会显著影响体系的能量和位错芯的电子结构,有利于改善NiAl的力学性能。
The authors have declared that no competing interests exist.

/
| 〈 |
|
〉 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}