
陶辉锦 , 周珊
, 周珊
TAO Huijin, ZHOU Shan
文献标识码: TG111
文章编号: 0412-1961(2017)06-0751-09
通讯作者:
收稿日期: 2016-10-20
网络出版日期: 2017-06-20
版权声明: 2017 《金属学报》编辑部 《金属学报》编辑部
基金资助:
作者简介:
作者简介 陶辉锦,男,1976年生,副教授
展开
摘要
采用Wagner-Schottky点缺陷热力学模型和第一性原理平面波赝势方法,计算研究了D019-Ti3Al金属间化合物中空位和反位2种类型点缺陷的形成焓、平衡浓度及相互作用。结果表明,这些缺陷的平衡浓度均随温度升高而增大,反位缺陷浓度均高于空位缺陷,Ti原子空位的浓度高于Al原子空位。在理想化学计量比成分下,Ti原子反位与Al原子反位缺陷浓度基本相当;在略偏离计量比的富Ti成分端,Ti原子的反位缺陷浓度高于Al原子;在富Al成分端则情形相反。计算结果表明,3种点缺陷对(AlTi-TiAl、TiAl-TiAl、VAl-AlTi)在基体中具有较强的聚集趋势,而其它类型的点缺陷对则有向基体扩散的趋势。
关键词:
Abstract
The intermetallics D019-Ti3Al has low specific density and high thermal resistance for both bulk and coating applications in engineering area. The point defects such as thermal vacancy, compostion vacancy and antisite defect have great influence on the properties of D019-Ti3Al, but are usally neglected. According to available research data from both theory and experiment, it is commonly considered that the thermal vacancies in D019-Ti3Al provide paths for atomic migration and diffusion, the antisite defects play an important role in the disordering of D019-Ti3Al, and the interaction between composition vacancy and antisite defect may have important influence on atomic diffusion and dislocation movement. So it is necessary to explore the mechanism of interaction between composition vacancy and antisite defect for more accurate understanding of the atomic diffusion, dislocation movement and plastic deform in D019-Ti3Al. In this work, the formation enthalpy, equilibrium concentration, and binding energy of composition vacancy and antisite defect in D019-Ti3Al intermetallics were calculated by using both the Wagner-Schottky model of point defect thermodynamics and the plane wave pseudopotential method in first-principles. Results suggest that, in the whole composition range of interest, the point defect concentrations increase with the increase of temperature. In particular, the concentrations of antisite defects are higher than those of vacancies, and the vacancy concentration of Ti is higher than that of Al. At the stoichiometric composition, the concentrations of antisite defects of Ti and Al are very close. At the Ti-rich side of component, the antisite defect of Ti dominates in concentration, while at the Al-rich side, that of Al dominates in concentration. For the calculated results of 3 types of point defect pairs, AlTi-TiAl, TiAl-TiAl and VAl-AlTi, they may have the strong trend to aggregate, while others may show the tend to diffuse into the matrix.
Keywords:
TiAl系金属间化合物,包括Ti3Al、TiAl和TiAl3等,因具有极高的抗氧化性、较高的比熔点和较低的比重而成为研究热点。特别是Ti3Al金属间化合物,因具有D019型晶体结构和低比重、高热阻等优良性质,不论是作为块体材料还是涂层材料,在工程领域均有着较强的应用需求,因此,对其成分、结构与性能之间相关性的研究也在不断深入[1,2]。近年来,Liu等[3,4]采用第一性原理和分子动力学方法,研究了动力学剪切变形诱发Ti3Al从hcp向fcc的结构转变,预测在剪切变形时双相合金中Ti3Al倾向于向TiAl发生结构转变。Yoo等[5]基于第一性原理局域密度泛函方法,研究了Ti3Al的相稳定性和体性质,发现Ti3Al基态的弹性常数、Young's模量和剪切模量比TiAl要高。Karkina等[6]采用嵌入原子势,通过分子动力学模拟研究了Ti3Al的位错芯结构和变形行为。Fu等[7]采用第一性原理全势线性缀加平面波方法,分别计算了Ti3Al的弹性常数、反相畴界和复合堆垛层错能以及Ti3Al/TiAl界面的层错能,发现Ti3Al中的化学键合与金属Ti中多中心键合相互作用类似,Al原子的存在加强了这种键合,使Ti3Al表现出更高的弹性模量和非常高的反相畴界能,且由于长程键合作用受到破坏,Ti3Al/TiAl界面的反相畴界和层错能比其组成相都要低。Wang等[8]采用第一性原理投影缀加波方法,计算研究了α2-Ti3Al (0001)表面能和γ-TiAl (111)/α2-Ti3Al (0001)界面的界面能,发现Ti3Al (0001)面比TiAl (111)面有更高的表面能和更大的表面弛豫,界面沿着Ti3Al (0001)面的分离功比TiAl (111)面的大。Xie等[9]采用嵌入原子势,对Ti3Al在快速凝固过程中纳米晶的形成,以及纳米晶Ti3Al在拉伸变形过程中的变形行为进行了研究,发现当冷却速率为1011 K/s时,纳米晶完全晶化,而且在快速凝固过程中形成的共格孪晶界,可能成为纳米Ti3Al合金的裂纹源。朴英锡等[10]应用固体与分子经验电子理论计算了Ti3Al及加入Nb后各相的价电子结构,并从均匀变形因子、解理能及位错行为等方面分析了Nb对Ti3Al脆性的影响,发现Nb使该合金的均匀变形因子及解理能提高,同时也减弱了Ti-Ti共价键,增加了其基面滑移,使得Ti3Al脆性有本质改善。Chen等[11]采用高分辨电镜和X射线能谱仪研究了高Nb含量TiAl合金中变形诱发的α2向γ相的转变。Al-Kassab等[12]采用真空感应悬浮熔炼方法制备了掺Nb TiAl/Ti3Al金属间化合物,并采用场离子显微镜、层析原子探针、透射电镜对其进行了结构分析,发现添加原子分数为5%的Nb后,双相合金结构以及合金的片层结构并未发生显著改变,TiAl相中的Nb原子优先占据Ti原子位置。Wei等[13]采用第一原理方法研究了双相TiAl-Ti3Al合金中O元素从原子占位到对界面能的影响,发现O元素在TiAl合金中的占位趋势由高到低分别为α2-Ti3Al、γ-α2相界面及γ-TiAl相,它的存在使得TiAl/Ti3Al界面的分离功降低了4%。Zhang等[14]研究了碳化物增强Ti3Al复合材料,发现Ti3AlC的硬度和弹性模量略高于Ti3Al,但远低于TiC,它能充当过渡层的角色来减少该复合材料在服役过程中的应力集中。Bratanich等[15]研究了Ti3Al作为储氢和热阻材料在吸氢过程中的化学反应和结构变化,发现氢在Ti3Al中的溶解导致了其晶体结构的无序化,氢与Ti3Al中的Ti元素反应,会导致不同相的产生。
以上研究主要针对Ti3Al组织结构与宏观性能的相关联系,但研究中发现尽管点缺陷对其性质影响很大,却常常被忽视。Rüsing等[16]采用Ti同位素原子44Ti作为示踪原子,首次研究了有序Ti3Al合金中的扩散行为,发现Ti在有序Ti3Al合金中的自扩散系数比在纯Ti中低,符合Arrhenius扩散行为,特别是在理想化学计量比成分的合金中没有明显的自扩散行为。Ti3Al相中的原子迁移是通过热空位进行的,无迹象表明有成分空位的形成和参与。Shirai等[17]采用正电子湮灭寿命谱研究了受辐照后的Ti3Al中的空位及其团簇,在该材料中没有检测到成分空位,所有的成分点缺陷均为反位缺陷。受辐照的材料在250 K发生回复,可归因于Ti空位的迁移,在350 K以上发生的回复,可归因于Ti和Al空位的迁移。Mishin等[18]采用嵌入原子势方法和周期边界条件,计算了Ti3Al中的点缺陷形成能和1200 K的点缺陷平衡浓度,发现反位缺陷占据主导地位,说明Ti3Al是反位无序的化合物,这与实验、Bragg-Williams模型和第一性原理计算的结果一致。Semenova等[19]基于平均场近似和统计热力学模型,计算比较了具有D019结构的非化学计量比Ti3Al中4种点缺陷的形成能,所预测的空位浓度很低,进一步预示着化学计量比的偏离几乎全部来自反位缺陷的贡献。Fröbel等[20]研究了Ti3Al的应变时效,发现在150~550 ℃的中温区间Ti3Al出现不连续的屈服和静态应力时效现象,这些现象取决于合金成分对化学计量比的偏离,且有可能是由Ti和Al的反位缺陷导致的。反位与空位缺陷的组合体可能形成反结构桥,从而为扩散提供了通道;在位错应力场中还可能发生重新取向,降低了位错的可移动性,从而影响合金的变形性能。
总结以上对D019-Ti3Al本征点缺陷的研究结果,可以得出以下几点共识:热空位为原子迁移和扩散提供了通道,成分空位很难从实验上检测到,反位缺陷是主要缺陷形式,对D019-Ti3Al无序化有重要作用,而且空位与反位缺陷的相互作用可能对原子扩散和位错运动有重要影响。因此,进一步研究Ti3Al中成分空位、反位缺陷及其与空位缺陷的相互作用机理极有必要,这将有助于更准确地理解和认识Ti3Al中原子扩散、位错运动和塑性变形行为。本工作采用第一原理计算研究方法,结合Wagner-Schottky点缺陷热力学模型,考察D019-Ti3Al中各种主要点缺陷的形成焓,预测这些点缺陷的浓度随温度和成分的变化规律以及相应点缺陷的形成激活能,进一步评估成分空位与反位点缺陷之间的可能相互作用,从而为D019-Ti3Al金属间化合物的实验制备和结构改造等提供有益的理论基础。
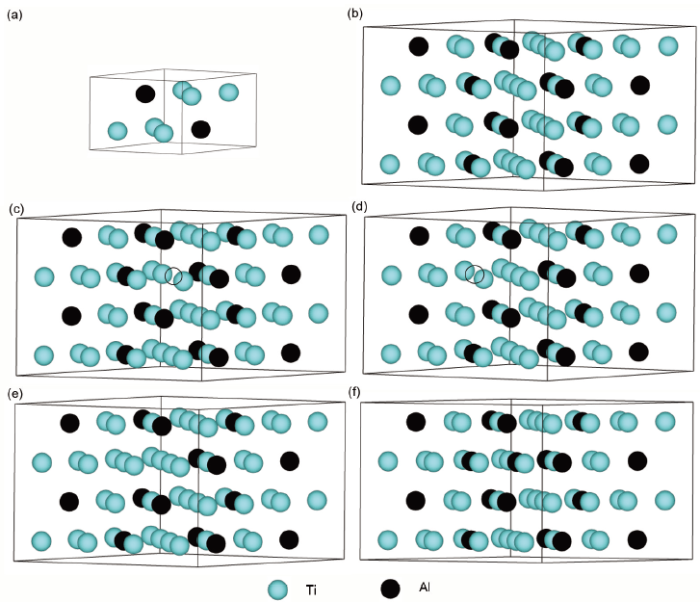
本工作所有计算基于第一原理平面波赝势方法。电子与离子之间的相互作用用投影缀加波方法(PAW)[21]来精确描述,电子之间的交换关联势采用广义梯度近似下的Perdew-Burke-Ernzerhof (PBE)方法来描述[22],Ti的3p4s3d和Al的3s3p电子作为Ti和Al的价电子,其余内层电子作为芯电子来考虑。晶胞模型简约Brillouin区的K点网格采用Monkhorst-Pack[23]方法来划分,系统总能量的计算采用Blöchl等[24]修正的Linear-Tetrahedron方法。电子Kohn-Sham波函数用平面波基组展开,其数目由经过收敛性测试后的动能截断点来确定,在计算精度确定为0.001 eV的条件下,Ti、Al及Ti3Al超胞能量的动能截断点确定为400.0 eV,计算采用VASP (Vienna Ab-inito simulation package)总能计算程序进行。在计算含点缺陷的晶胞结构时构建了一个含有64个原子的超胞,倒易空间网格的划分取为2×2×3。图1给出了D019-Ti3Al金属间化合物及含有各种点缺陷的超胞结构示意图。
计算得到的D019-Ti3Al金属间化合物的晶格常数a=0.5757 nm,c=0.4657 nm,c/a=0.809;弹性模量为114.6 GPa,与文献[3,5,6]中的计算值和文献[25,26]中的实验值符合较好。
Wagner-Schottky模型[27]可以用来研究稀浓度的点缺陷随成分和温度的变化关系。本工作定义点缺陷浓度时采用原子浓度,即质点i (原子或空位)在α 亚点阵位置的原子浓度
其中,
其中,
含缺陷D019-Ti3Al合金的形成焓可按下式计算:
式中,m和n是晶胞中所含原子数目;
其中,
图1 D019-Ti3Al金属间化合物空位和反位点缺陷晶胞模型示意图
Fig.1 Schematics of supercell models of D019-Ti3Al intermetallics
(a) conventional cell (b) perfect supercell (c) supercell with Ti vacancy (d) supercell with Al vacancy (e) supercell with Ti antisite defect (f) supercell with Al antisite defect
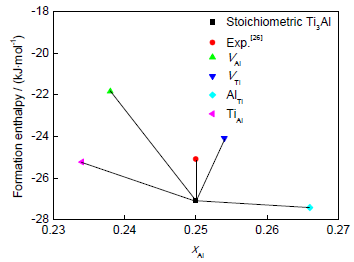
根据式(3)和(4)可计算得到包含点缺陷(VAl、VTi、AlTi、TiAl)的D019-Ti3Al的形成焓,结果如图2所示。根据热力学可知,较低的形成焓意味着更加稳定的状态,从图2可以看出,含反位缺陷D019-Ti3Al的形成焓比空位缺陷的更低,表明反位缺陷在能量上可能是D019-Ti3Al中相对稳定的缺陷形式。本工作计算所得理想D019-Ti3Al的形成焓与文献[26]中报道的实验值相对误差小于8%,符合较好。
图2 含有单个点缺陷的D019-Ti3Al形成焓与Al原子成分(xAl)之间的关系
Fig.2 Relations between formation enthalpy and atomic content of Al (xAl) in D019-Ti3Al with single point defect (VAl—Al vacancy, VTi—Ti vacancy, AlTi—antisite Al, TiAl—antisite Ti)
根据式(2)和图2的形成焓结果,得到以理想D019-Ti3Al为参考态的单个点缺陷(VAl、VTi、AlTi、TiAl)形成焓Hd[28]:
计算得到的空位和反位缺陷形成焓结果与其它计算结果和实验值对比见表1。
由表1可知,本工作计算结果与文献[19,29]中报道过的计算结果有差异,这可能是计算模型和方法的不同所引起的,但总的变化趋势基本一致,D019-Ti3Al中空位形成焓均大于反位形成焓,意味着反位缺陷更容易形成。同时,本工作计算结果表明:Al空位(VAl)的形成焓大于Ti空位(VTi),Ti反位(TiAl)的形成焓值要大于Al反位(AlTi),这意味着对于偏离理想化学计量比的富Al型Ti3Al化合物,其可能的主要结构缺陷形式为AlTi和VTi,且AlTi的形成焓远小于VTi,AlTi的浓度将占绝对优势。同时,对于富Ti型Ti3Al化合物,可能出现的主要点缺陷形式为TiAl和VAl,且TiAl缺陷的浓度将占明显优势。总之,在偏离化学计量比的D019-Ti3Al金属间化合物中,总是更容易形成反位缺陷,富Ti或者富Al型D019-Ti3Al将分别以TiAl或者AlTi为主要的点缺陷形式存在。
在计算Ti3Al金属间化合物点缺陷浓度时需要考虑温度的因素,并且不能忽略熵对系统的影响。简化起见,如果仅考虑组态熵的影响,可以通过平均场近似[27,28]来计算组态熵:
式中,
考虑到D019-Ti3Al的相区成分
为进一步研究点缺陷浓度与温度之间的关系,选取图3中满足化学计量比条件,即
表1 D019-Ti3Al金属间化合物空位和反位形成焓
Table 1 Calculated formation enthalpies (H) of vacancies and antisite defects in D019-Ti3Al (eVatom-1)
| Work | ||||
|---|---|---|---|---|
| Present work | 3.4272 | 1.9719 | -0.2093 | 1.2416 |
| Cal.[19] | 1.5 | 1.5 | 0.6 | 0.6 |
| Cal.[29] | 2.76 | 3.15 | 0.54 | 0.16 |
| Exp.[30] | - | 1.55 | - | - |
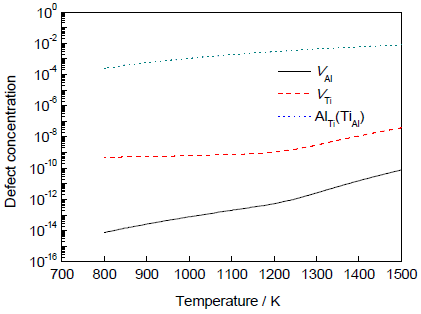
图4已经给出了计算预测的理想化学计量比D019-Ti3Al中不同点缺陷浓度随温度的变化趋势。在实验研究中,实际测定点缺陷形成激活能通常都基于Arrhenius方程[28],即:
其中,c为点缺陷浓度,A为平衡常数,Q为点缺陷形成激活能(kJ/mol),R是摩尔气体常数,T为热力学温度。对式(8)取以10为底的对数,可得:
可见,
计算结果如表2所示,该表给出了在不同温度区间进行拟合的结果。所有的拟合结果表明:VAl缺陷的形成激活能最大,VTi次之,AlTi和TiAl的形成激活能最小且相当。表2所预测的形成激活能实质上是一种表观激活能,可直接与实验结果对比。由表2可知,800~1500 K全部温度范围拟合的结果比分段拟合的结果更加接近文献[18]的计算和所提供的实验结果。
图3 在不同温度下D019-Ti3Al金属间化合物的点缺陷浓度与成分之间的关系
Fig.3 Point defect concentrations vs composition (xAl) of D019-Ti3Al intermetallics at different temperatures
(a) 873 K (b) 1073 K (c) 1273 K (d) 1473 K
图4 理想化学计量比D019-Ti3Al点缺陷浓度与温度的关系
Fig.4 Point defect concentration as a function of temperature in stoichiometric D019-Ti3Al
图5 D019-Ti3Al金属间化合物点缺陷浓度(c)与温度(T -1)之间的关系
Fig.5 Point defect concentrations (c) as a function of temperature(T -1) in D019-Ti3Al intermetallics
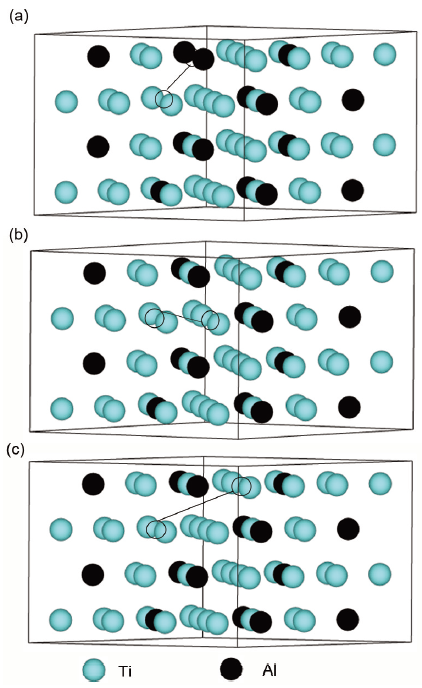
图6 D019-Ti3Al中第一、第二和第三近邻VAl-VTi点缺陷对的晶胞模型示意图
Fig.6 Schematics of supercell model of the first (a), second (b) and third (c) nearest VAl-VTi point defect pair in D019-Ti3Al
表2 D019-Ti3Al中空位和反位缺陷形成激活能的实验值预测
Table 2 Prediction of activation energy (
| Temperature | ||||
|---|---|---|---|---|
| 800~1500 K | 1.489 | 0.762 | 0.511 | 0.511 |
| 800~1200 K | 0.866 | 0.139 | 0.513 | 0.513 |
| 1200~1500 K | 2.944 | 2.218 | 0.508 | 0.508 |
| Cal.[18] | 1.805 | 1.314 | 0.313 | 0.313 |
| Exp.[18] | 1.55 |
基于以上计算和预测,可以研究单个点缺陷(VAl、VTi、AlTi、TiAl)之间的可能相互作用。现将这些点缺陷进行组合,得到10种可能的缺陷对(d-d')结构,即VAl-VTi、VAl-VAl、VAl-TiAl、VTi-VTi、VTi-TiAl、VAl-AlTi、TiAl-TiAl、VTi-AlTi、AlTi-TiAl、AlTi-AlTi。同时,根据点缺陷之间的距离进行了从第一近邻到第三近邻的超胞能量计算,图6以双空位点缺陷对(VAl-VTi)为例给出了这些第一、第二和第三近邻点缺陷对的超胞模型。根据这些模型计算这些点缺陷对的总能量,并与单个点缺陷进行能量对比,可以评估它们以理想D019-Ti3Al为参考态的形成焓和结合能,从而预测它们之间的吸引或排斥作用[31]。同样的,以双空位点缺陷对(VAl-VTi)为例,其第一、第二和第三近邻点缺陷对的形成焓Hd-d'和结合能Fd-d'计算公式如下[32]:
式中,
表3 D019-Ti3Al中最近邻点缺陷对的形成焓和结合能
Table 3 Formation enthalpy (Hd-d') and binding energy (Fd-d') of the nearest point defect pairs in D019-Ti3Al
| Point defect pair | Hd-d' / eV | Fd-d' / eV | |||||
|---|---|---|---|---|---|---|---|
| First nearest | Second nearest | Third nearest | First nearest | Second nearest | Third nearest | ||
| VAl-VTi | 5.5203 | 5.5045 | 5.9087 | -0.1212 | -0.1054 | -0.5096 | |
| VAl-AlTi | 3.1869 | 3.3116 | 3.4959 | 0.0310 | -0.0937 | -0.2780 | |
| VTi-TiAl | 3.6256 | 3.5722 | 3.5010 | -0.4121 | -0.3587 | -0.2875 | |
| AlTi-TiAl | 0.8544 | 1.0014 | 1.0454 | 0.1779 | 0.0309 | -0.0131 | |
| VAl-TiAl | 4.8687 | 4.7788 | 4.9194 | -0.1999 | -0.1100 | -0.2506 | |
| VTi-AlTi | 2.0442 | 2.1019 | 1.9564 | -0.2816 | -0.3393 | -0.1938 | |
| VAl-VAl | 7.4272 | 7.4542 | 7.5333 | -0.5728 | -0.5998 | -0.6789 | |
| VTi-VTi | 4.4483 | 4.4546 | 4.3857 | -0.5045 | -0.5108 | -0.4419 | |
| AlTi-AlTi | -0.2922 | -0.2903 | -0.5427 | -0.1264 | -0.1283 | 0.1241 | |
| TiAl-TiAl | 2.4479 | 2.5269 | 2.5390 | 0.0353 | -0.0437 | -0.0558 | |
由表3结合能Fd-d'结果可知:(1) 结合能随点缺陷对距离的增加呈现出与焓变化相反的趋势;(2) 对于结合能为正值的点缺陷对,它们能够稳定存在,其中聚集在最近邻位置的点缺陷对有AlTi-TiAl、TiAl-TiAl、VAl-AlTi,聚集在第二近邻位置的点缺陷对有AlTi-
TiAl,聚集在第三近邻位置的点缺陷对有AlTi-AlTi;(3) 在最近邻配对的点缺陷对中,结合能大小顺序为AlTi-TiAl>TiAl-TiAl>VAl-AlTi,这表明,AlTi将更倾向选择TiAl而非VAl与其聚集,TiAl将更倾向选择AlTi而非TiAl与其聚集,但其余的最近邻点缺陷则不会聚集,而是相互排斥,向基体分散和扩散。
(1) D019-Ti3Al金属间化合物中Al和Ti的原子反位缺陷形成焓绝对值更大,相对空位缺陷更为稳定;所有点缺陷的浓度均随温度升高而增加,且反位缺陷浓度始终大于空位缺陷,是该金属间化合物中最主要的点缺陷形式。
(2) 在整个D019-Ti3Al相区成分范围内,Al空位浓度和Ti反位缺陷浓度均随Al的原子分数增加而下降,但Ti空位和Al反位浓度变化趋势相反;在理想化学计量比(Al原子分数为25%)处,Ti反位与Al反位缺陷浓度基本相当,但在富Ti端,Ti反位浓度高于Al反位浓度,在富Al端,Al反位浓度高于Ti反位浓度。
(3) 基于Arrhenius方程计算预测了各种点缺陷的形成激活能,发现Al空位形成激活能最大,Ti空位次之,Al反位和Ti反位形成激活能较小且数值相当;通过对不同点缺陷之间结合能计算的考察,发现点缺陷对AlTi-TiAl、TiAl-TiAl和VAl-AlTi点缺陷,特别是AlTi-TiAl之间存在很强的相互吸引,容易聚集,其余的点缺陷对则存在着相互排斥,有向基体分散与扩散的趋势。
致谢 感谢中南大学高性能计算中心和江勇教授课题组为所有计算工作提供的支持和帮助。
The authors have declared that no competing interests exist.

/
| 〈 |
|
〉 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}