
蒋宗佑
JIANG Zongyou
通讯作者:
收稿日期: 2016-05-27
网络出版日期: 2016-01-11
版权声明: 2017 《金属学报》编辑部 《金属学报》编辑部
基金资助:
作者简介:
作者简介: 蒋宗佑, 男, 1994年生, 硕士生
展开
摘要
采用密度泛函方法研究了水分子在Au, Cu和AuCu二元金属合金表面的不同吸附状态和裂解反应路径, 并且比较不同表面的催化性能. 结果表明, 研究中所考虑4种模型的反应活性顺序如下(以反应活化能为比较标准): Au(111) < AuCu(111)-Cu < AuCu(111)-Au < Cu(111). 相对于AuCu(111)-Cu表面和Au(111)表面, 这与水分子的分子吸附状态在AuCu(111)-Au表面和Cu(111)表面的吸附能相对较小, 而解离吸附状态时的吸附能相对较大有关. 根据金属催化剂表面与吸附物的电子转移和成键电子结构, 认为电子转移越多, 与表面的相互作用越强; 而金属原子的d电子态与水分子1b1电子态或者裂解产物的类1b1电子态之间的重叠、杂化程度决定了两者之间相互作用的强弱. 在实际反应体系中, 用AuCu二元金属合金替代贵金属Au催化剂不仅可以降低材料成本, 还可以提高反应活性.
关键词:
Abstract
In order to reduce the amount and cost of gold catalyst in practical application, it is an effective technical strategy to construct binary metal alloy with gold and transition metals. In this work, the adsorption behaviors and dissociation reaction path of water on the different surfaces of Au, Cu, and AuCu binary alloy were studied by using DFT calculations. Based on the calculations, the corresponding catalytic performance of each model was further analyzed. The calculated results showed that the catalytic activity of the considered four surface models is on the following order: Au(111) < AuCu(111)-Cu < AuCu(111)-Au < Cu(111), if using the active energy as comparison standard. The underlying reason of this phenomenon is closely related with the adsorption behavior: the molecular adsorptions of water on Cu(111) and AuCu(111)-Au surfaces have relatively small adsorption energies; while at the same time, the dissociation adsorptions of water on these two surfaces have relatively large adsorption energies. Based on the electron transfer and bonding electronic structure, it could be found that the more electrons transfer between surface and water or H+OH groups, the more strong interaction between catalyst and adsorbate. Furthermore, the overlapping or hybridization between the d states of metal atoms on the surface and the 1b1 states of water or lb1-like states of H+OH groups also determines this interaction. Therefore, using AuCu binary alloy to replace Au as catalyst or co-catalyst reduces cost, and enhances the catalytic activity.
Keywords:
Au作为一种具有化学惰性的贵金属, 广泛应用于珠宝首饰及微电子集成电路等领域. 作为一种催化剂, 对Au的研究最早可追溯到1926年Au催化H2+O2的反应, 但是Au的催化反应速率比Ag的要低得多[1]. 1985年, Hutchings[2]在研究乙烯氢氯化反应机理时, 曾预测负载型Au催化剂是该反应中催化活性最高的催化剂, 但是这一结论直到1988年才被实验证实. 1987年, Haruta等[3,4]报道超细Au颗粒显示出极高的CO低温氧化活性. 虽然Au的催化活性较低, 但是其本身具有很高的催化选择性[5], 因此Au催化剂大多数情况下应用于选择性氧化反应和有机反应.
Au在催化领域中最主要的3种应用形态是: 负载型Au颗粒、Au薄膜和Au纳米颗粒. 2015年, Wu等[6]发现高价氧化态的Au3+有极强的亲氧能力, 可取代强氧化剂, 并且其前驱体很容易制备且易于保存, 这是催化领域中的一个重大发现. 乔波涛等[7]通过简单的沉淀吸附法制备Co3O4负载Au单原子催化剂(Au1/Co3O4), 发现Au负载量仅为0.05% (质量分数)即可在室温条件下实现CO的完全转化, 并提出了“单原子催化”的概念, 表明负载型Au单原子在CO氧化方面具有独特的催化作用. 除了自身具有的催化活性之外, 纳米Au粒子还可以作为助催化剂负载在光催化材料表面, 可增加光生电子-空穴对的分离效率, 从而提高光催化材料的活性. 例如, 代卫炯等[8]用光沉积法在TiO2载体上负载Au, 发现在Au和TiO2界面处形成的Schottky势垒, 有效阻止了光生电子与空穴的复合. 相比于没有Au助催化剂存在的情况, 前者的光催化活性得到了大幅提高. 由于负载型Au催化剂的独特性能, 负载型Au催化剂催化CO低温氧化的反应仍然是目前的研究重点. 但是, Au本身价格昂贵, 资源有限, 且其催化活性也有待提高, 因此需要引入新的思路, 在保持其催化活性的前提下减少Au的用量.
为了降低催化剂成本并尽可能提高催化活性, 在实际应用中通常考虑以非贵金属替代部分贵金属. 相关研究[9~12]表明, 过渡金属Cu是最有希望的非贵金属催化剂之一. 在催化领域中, Cu主要应用在有机反应中的催化方面, 包括催化偶联反应、催化多组分反应和Cu金属配合物等应用. 1901年, Ullmann和Bielecki[13]首次报道了Cu催化的交叉偶联反应, 即2个芳基直化物之间形成C—C键的偶联反应, 开创了Cu在催化偶联反应方面的先河. Nadler和Sanz[14]采用第一性原理分子动力学模拟H2O在Cu(111)表面的作用, 发现Cu表面的存在将改变H键网络, 并使H—O键加强, 这种影响类似于将纯H2O加热所带来的结果. 2016年, Shao等[15]报道了在Cu催化不对称炔丙基转化研究中, 通过运用一种脱硅活化的新方法, 成功实现了Cu催化的炔丙醇酯与β-萘酚及富电子苯酚间的不对称[3+2]环加成反应. 2009年, 厉嘉云等[16]报道了N-杂环卡宾Cu配合物的应用前景及其催化机理. 由于纳米技术的迅猛发展, 研究人员发现包括Cu及其化合物在内的纳米材料具有很多新的特点, 比如催化活性高、应用范围广、环境友好、价格低廉、能多次循环利用等. 2014年, Manthiram等[17]发现将Cu纳米粒子负载于玻璃碳上面(n-Cu/C)并用于CO2的甲烷化反应, 其效率比单独使用高纯度Cu箔电极高了4倍. Kundu和Pradhan[18]发现纳米结构的CuS对亚甲基蓝降解有着较高的效率. 此外, 工业上的骨架Cu催化剂主要作为丙烯腈水合制丙烯酸胺反应的催化剂, Cu系催化剂在CO转化及某些典型的加氢还原反应中具有较高的活性. 鉴于Cu在催化领域的诸多优点, 以及其成本的低廉与储量的丰富, 研究者希望找到一种将Au的特点与Cu的优点相结合的有效途径.
研究[19]发现, 金属与金属之间形成的双/多金属结构, 其物理化学性能往往优于单金属, 这种现象被称之为金属之间的协同效应. 由于催化剂的表面结构是决定其催化性能的关键因素, 而引入第二种金属又可以改变单金属作为催化剂时的表面结构, 因此可以将2种或多种金属通过不同比例、不同结构与不同组合的方法制备成具有较好催化性能的双/多金属催化剂. 而Au-Cu作为贵金属体系中重要的基础合金体系, 在催化领域中, 不仅降低了Au单独作为催化剂时的成本, 也提高了Cu或Au单独存在时的催化性能. 2015年, Fiorenza等[20]在挥发性有机化合物的氧化和CO选择氧化实验中发现, 在低温下, Au-Cu/CeO2比Au/CeO2或Cu/CeO2拥有更高的CO2产量. 2012年, 李力成等[21]制备出负载型双金属催化剂Au-Cu/TiO2, 并用于CO催化氧化反应中. 发现Cu可以提高Au催化剂的催化性能, Au-Cu/TiO2催化剂比Au/TiO2和无孔TiO2负载的Au催化剂具有更好的CO催化氧化稳定性. 这可能与Au, Cu合金化和TiO2的介孔结构有关.
尽管关于Au-Cu合金催化剂的实验报道较多, 但对其相关的理论研究还比较少, 迫切需要从理论上研究其催化反应的机理, 以解释现有的实验现象并指导后期的研究工作. 不论是均相催化、非均相催化或者其它类型的催化反应, 催化过程大都发生在催化材料的表面, 所以反应物分子的吸附分解是催化反应的必经步骤, 即催化反应的机理与反应分子在催化剂表面的吸附行为是不可分割的.
本工作以Au, Cu及其合金为研究对象, 通过理论模拟与计算, 揭示其催化的微观机制. H2O是最常见的溶剂, 水分子本身就可以参与分解反应产生O2和H2, 且水分子结构简单, 同时它可以通过自身的解离产物(如表面羟基、羟基自由基等是一些催化反应的活性物种)影响后续的催化反应, 因此将水分子作为研究催化机理的理想探针分子. 本工作从微观角度, 通过理论计算, 模拟催化过程中H2O在Au, Cu和Au-Cu合金表面的吸附与分解, 研究其吸附状态、成键机理和活性吸附位点等, 并据此分析Au, Cu和Au-Cu合金催化剂的反应机理.
本研究所有理论计算工作均由Materials Studio软件中的CASTEP (Cambridge serial total energy package)模块完成. 采用平面波超软赝势方法来描述电子-离子实间的相互作用, 以保证在一定计算精度的前提下节省计算时间和计算资源. 电子波函数通过平面波基组展开, 其中平面波截断能设置为380 eV. 电子-电子间相互作用的交换关联能由广义梯度近似(generalized gradient approximation, GGA)中的PBEsol泛函进行描述, 这是目前对固体材料较为准确的理论计算方法. 几何结构优化收敛标准设置如下: 2次相邻离子步之间的总能量差小于1.0×10-6 eV/atom, 原子间相互作用力小于0.1 eV/nm, 原子所受应力小于0.02 GPa, 原子位移小于5.0×10-5 nm. 自洽场运算中电子步的能量收敛标准为5.0×10-7 eV/atom. 其它相关设置如下: K-points设置为2×2×1, 快速Fourier变换(FFT)的网络设置为54×54×360. 为了得到更加准确的计算结果, 所有计算均采用偶极校正, 以消除表面不对称所导致的影响.
Au和Cu的原始模型采用标准fcc结构(空间群为
对于水分子的分解过程, 本工作只分析对比裂解的初始阶段, 即只有一个O—H断裂的情形. 在对不同可能的分子吸附和解离吸附构型进行几何结构优化、并得到最稳定的吸附构型之后, 以分子吸附作为反应物, 以解离吸附作为反应产物. 然后采用完全线性同步转变/二次同步转变(complete linear synchronous transit/quadratic synchronous transit, Complete LST/QST)方法搜索反应物和反应产物之间的过渡态, 其中: RMS收敛设置为0.1 eV/nm, 最大QST步数设置为5. 在得到初始过渡态之后再次进行过渡态确认, 其收敛标准设置为: 总能量差小于5.0×10-6 eV/atom, 原子间相互作用力小于0.2 eV/nm, 原子位移小于3×10-4 nm, 最大的镜像数设置为10.
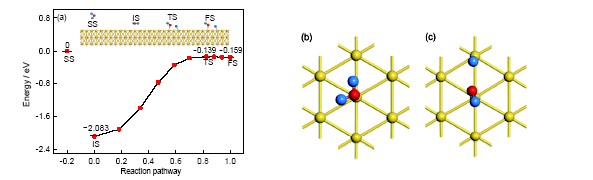
Au的密排面(111)晶面是其作为催化材料研究最多的对象[22]. 本计算中首先以水分子作为研究对象, 分析其在Au(111)晶面的吸附与分解行为, 计算结果如图1a所示, 水分子在此表面上的分子吸附状态和解离吸附状态对应的吸附能分别为2.083和0.159 eV. 即从水分子到H+OH的分解过程是吸热反应, 对应的反应热为1.924 eV. 也就是说, 水分子在Au(111)晶面吸附时, 分子吸附状态在能量上更占优势, 在一般情况下水分子在此晶面上以分子吸附为主. 相关的结构参数列于表1. 结合表1和图1可发现, 水分子在分子吸附状态时, 水分子的分子平面与Au(111)晶面接近于平行(夹角约为4.375°), 2个O—H键的键长和键角与孤立水分子的键长和键角也基本相同, 只有稍许变化, 说明表面对水分子的微观结构影响很小, 这可能与该表面上H2O的微观结构比较稳定有关. 值得注意的是, 水分子在此状态下, 2个H原子几乎完全对称地指向邻近的2个Au原子, 这种相互作用使其保持了与孤立分子基本相同的结构. 解离吸附时H+OH基团的平面与Au(111)晶面的夹角是57.029°, 未裂解的O—H键长与孤立水分子中的O—H键长相差很小, 裂解之后的O—H键长为0.2237 nm. 裂解后产生的H原子与邻近的Au原子成键, 两者之间的键长为0.1619 nm (这一键长只相当于分子吸附时H原子与对应Au原子之间距离的一半). 在2个状态之间反应路径上的鞍点(即过渡态)对应的吸附能为0.139 eV, 在此状态H+OH基团的平面与Au(111)晶面的夹角是74.141°. 在反应路径上, 过渡态的结构是不稳定的, 它向前向后都很容易成为分子吸附或解离吸附状态. 从图1a中可以发现, 由于过渡态与终态(反应产物)存在一定的相似之处, 所以这一过渡态属于“晚垒”. 从能量角度来看, 以分子吸附为初始态、解离吸附为终态的反应活化能为1.944 eV, 其逆反应的活化能为0.020 eV, 即在Au(111)晶面上水分子分解反应的逆反应更容易进行, 所以在此表面上水分子以分子吸附为主要的存在形态. 分子吸附与解离吸附的局部示意图分别如图1b和c所示. 可以看出, 2种吸附状态都是顶位吸附, O原子与其所吸附的Au原子的距离分别为0.2571和0.2113 nm.
图1 水分子在Au(111)表面上不同的吸附状态与反应路径, 及分子吸附和解离吸附的局部构型
Fig.1 Different adsorption states and reaction path of water on Au(111) surface (a), and the local configurations of molecular adsorption (b) and dissociation adsorption (c) (SS—separated state, IS—initial state, TS—transit state, FS—final state)
表1 本工作计算得到水分子在不同材料表面吸附与解离的结构参数
Table 1 Structural parameters of water adsorption and dissociation on different materials surfaces in this work
| Model | State | |||||||
|---|---|---|---|---|---|---|---|---|
| Au(111) | IS | 0.0984 | 0.0985 | 104.587 | 0.3276 | 0.3185 | 0.2571 | 4.375 |
| TS | 0.0983 | 0.1883 | 154.758 | 0.3768 | 0.1641 | 0.2125 | 78.141 | |
| FS | 0.0986 | 0.2237 | 153.875 | 0.3664 | 0.1619 | 0.2113 | 57.029 | |
| Cu(111) | IS | 0.0988 | 0.0988 | 105.354 | 0.2857 | 0.2858 | 0.2168 | 15.190 |
| TS | 0.0987 | 0.1569 | 126.960 | 0.2778 | 0.1773 | 0.1968 | 41.649 | |
| FS | 0.0981 | 0.3866 | 95.969 | 0.2731 | 0.1735 | 0.1997 | 88.792 | |
| AuCu(111)-Au | IS | 0.0983 | 0.0985 | 105.055 | 0.3646 | 0.3135 | 0.2646 | 13.405 |
| TS | 0.0986 | 0.1503 | 141.136 | 0.2922a | 0.1839a | 0.2310a | 38.957 | |
| 0.2992b | 0.1915b | 0.2314b | ||||||
| FS | 0.0982 | 0.3077 | 95.809 | 0.1972a | 0.1762a | 0.1745a | 83.432 | |
| 0.2527b | 0.1727b | 0.1874b | ||||||
| AuCu(111)-Cu | IS | 0.0987 | 0.0988 | 105.907 | 0.3226 | 0.3053 | 0.2136 | 20.408 |
| TS | 0.0975 | 0.2211 | 127.245 | 0.2931a | 0.1624a | 0.1852a | 21.010 | |
| 0.3097b | 0.2483b | 0.2997b | ||||||
| FS | 0.0987 | 0.4212 | 114.274 | 0.2587a | 0.1914a | 0.1905a | 22.745 | |
| 0.2872b | 0.1755b | 0.2328b | ||||||
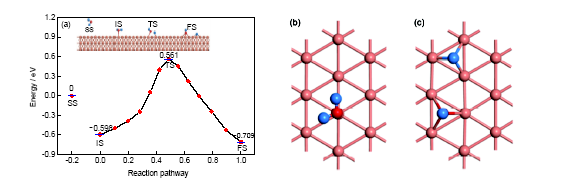
Cu(111)表面的催化性能也是受关注较多的课题之一[23,24]. 从表1及图2可以看出, 水分子处于分子吸附状态时, 情形与水分子在Au(111)表面的分子吸附状态类似: 水分子的键长、键角与孤立分子基本一致, 对称指向邻近的2个Cu原子, 分子平面几乎平行于表面. 但是在数值上有细微的差别, 如键长、键角稍大, 与表面的夹角相对较大(约为15.190°). 在裂解过程中, 水分子的分子平面逐渐竖立在表面上, 其中1个H原子被活化而与O原子分离, 吸附到邻近的空位上, 而裂解形成的OH基团则以O原子端朝下吸附最近邻的空位上, 成为表面吸附羟基, 形成最终的解离吸附状态. 在解离吸附状态下, H+OH基团的平面与表面几乎垂直(夹角约为88.792°), OH基团的氧原子和裂解的H原子与周围3个Cu原子分别形成等边四面体结构, O—Cu键长为0.1977 nm, H—Cu键长为0.1735 nm. 从反应路径上来看, 水分子在此表面上的解离吸附比分子吸附更占优势, 对应吸附能分别为0.709和0.598 eV. 也就是说, 水分子在Cu(111)表面上为分子吸附状态, 反应物的反应热为0.111 eV, 属于放热反应. 在势能面上, 初态(分子吸附)和终态(解离吸附)之间的过渡态的吸附能为-0.561 eV, 如果从分子吸附状态出发, 其反应活化能为1.159 eV; 而从解离状态出发, 其反应活化能为1.270 eV, 也就是说在Cu(111)表面, 水分子分解反应的正方向的反应更容易进行, 所以在此表面上水分子以解离吸附为主要的存在状态. 这说明对于水分子的裂解反应而言, Cu(111)表面具有较高反应活性.
图2 水分子在Cu(111)表面上不同的吸附状态与反应路径, 及分子吸附和解离吸附的局部构型
Fig.2 Different adsorption states and reaction path of water on Cu(111) surface (a), and the local configurations of molecular adsorption (b) and dissociation adsorption (c)
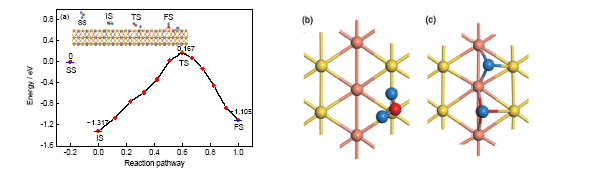
关于AuCu(111)表面的研究与应用已有诸多报道[25,26]. 本工作中分别优化了2种水分子可能的吸附构型, 即O原子位于Au原子上吸附(标记为AuCu(111)-Au), 以及O原子位于Cu原子上吸附(标记为AuCu(111)-Cu). 从图3及表1可以看到, 对于AuCu(111)-Au模型中的分子吸附状态, 水分子平面与表面接近于平行(夹角约为13.405°)吸附, 2个H原子分别指向邻近的Au和Cu原子, 对应的距离分别0.3587和0.4757 nm. 分子吸附时的吸附能为1.317 eV. 在解离吸附状态下, H+OH基团的平面与表面接近垂直(夹角约为83.432°), 裂解产物OH表面羟基竖直吸附于2个Cu原子之间的桥位, 另一裂解产物H原子吸附于由2个Cu原子和1个Au原子构成的空位. 解离吸附的吸附能为1.105 eV, 由此可见, 水分子在这个吸附位上的裂解反应是吸热反应, 对应的反应热为0.212 eV. 分子吸附和解离吸附之间的过渡态对应吸附能为0.167 eV, 其微观结构接近于终态, 所以属于“晚垒”过渡态. 从分子吸附状态出发, 反应活化能为1.484 eV; 从解离状态出发, 反应活化能为1.272 eV. 由于正反应活化能较高, 反应较难进行, 而且初态分子吸附的吸附能较高, 因此水分子在这个吸附位上以分子吸附为主要的存在形态. 分子吸附与解离吸附的局部示意图如图3b和c所示, O原子与其所吸附的Au原子的距离分别为0.2646 和0.2527 nm, 与最近邻Cu之间的距离分别为0.3935, 0.4076 和0.1972 nm.
图3 水分子在AuCu(111)-Au表面上不同的吸附状态与反应路径, 及分子吸附和解离吸附的局部构型
Fig.3 Different adsorption states and reaction path of water on AuCu(111)-Au surface (a), and the local configurations of molecular adsorption (b) and dissociation adsorption (c)
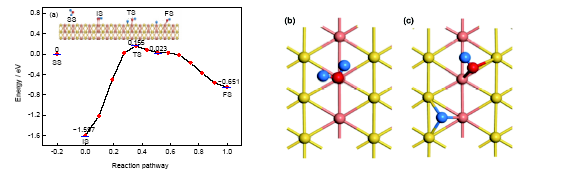
从图4和表1可以看出, 对AuCu(111)-Cu模型, 分子吸附时水分子平面与表面的夹角约为20.408°, 过渡态时的夹角约为21.010°, 解离吸附时H+OH基团的平面与表面夹角约为22.745°, 可见水分子或者裂解产物的平面与表面的夹角在裂解前后的变化较小. 在裂解过程中, 其中1个H原子被活化之后与O原子分离, 并逐渐靠近表面, 最终吸附于由2个Cu原子和1个Au原子构成的空位; 同时O原子偏离顶位吸附, 与其相连的H原子则远离表面(形成的表面羟基是倾斜在表面上的), 最终吸附于2个Cu原子之间的桥位. 从图4a可以看出, 水分子在此吸附位上分子吸附和解离吸附对应吸附能分别是1.597和0.651 eV. 也就是说, 水分子在此吸附位上裂解反应的反应热为0.946 eV, 是吸热反应. 分子吸附和解离吸附之间过渡态对应的吸附能为0.155 eV, 其微观结构接近于分子吸附状态, 属于“早垒”过渡态. 与以上其它3个反应路径不同, 在这一反应路径0.5附近有一个吸附能为0.023 eV的中间产物. 从总的反应路径来看, 从分子吸附初态出发, 水分子需要跃过1.752 eV的活化能势垒, 而其逆反应只需要0.806 eV. 这说明水分子在AuCu(111)的Cu位裂解时, 正反应较难进行, 所以在这一吸附位上水分子以分子吸附为主要的存在形状. 分子吸附与解离吸附的局部示意图如图4b和c所示, O原子与其所吸附的Cu原子的距离分别为0.2136和0.1905 nm.
图4 水分子在AuCu(111)-Cu表面上不同的吸附状态与反应路径, 及分子吸附和解离吸附的局部构型
Fig.4 Different adsorption states and reaction path of water on AuCu(111)-Cu surface (a), and the local configurations of molecular adsorption (b) and dissociation adsorption (c)
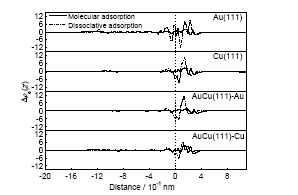
根据沿表面法线方向的平均电子密度变化(即平均差分电子密度曲线)可以分析在不同吸附状态下水分子或者裂解产物与表面之间的电荷转移情况. 从图5可以看出, 对Au(111)面上水分子的分子吸附而言, 表面附近的电子密度减小, 说明有部分电子传递到了水分子上. 同时在水分子与表面之间的区域当中, 2种吸附状态都是电子密度先增大后减小, 解离吸附对应的变化幅度更大. 对水分子或裂解产物而言, 其远离表面一端的电子密度减小, 说明不论是分子吸附还是解离吸附, 电子均集中分布在O原子附近. 对Cu(111)表面的吸附而言, 水分子吸附之后, 表面的电子密度变化很小, 更主要的变化是水分子或者裂解产物内部电子的重新分布, 导致电子主要集中在O原子; 在解离吸附状态下, 表面附近的电子大部分向水分子传递, 裂解产物中O原子位置附近的电子密度明显增加, 说明电子集中分布在这个区域. 对于AuCu(111)-Au模型吸附, 分子吸附时表面的电子变化情况并不明显, 而且表面上电子密度稍微有所增加; 解离吸附时表面上电子的情况出现先减小后增加、再减小的情况. 对于表面的电子变化很小的情况, 这可能主要是由于水分子内部电子的重新分布, 导致电子主要集中到O原子附近. 对于AuCu(111)-Cu模型吸附, 水分子吸附到表面之后, 表面电子密度略微增加, 说明在吸附过程中水分子的电子不仅有部分传递到表面上, 而且其内部也发生重组, 电子更加向其O原子靠近; 在解离吸附状态下, 电子先减小再增加, 而在分子吸附时则是一直增加. 可见对于分子吸附, 由于其与表面作用强于解离吸附, 分子吸附状态应该更加稳定. 综上所述, 电子转移的计算结果表明, 转移的电子数越多, 意味着吸附物与表面的相互作用越强, 因此在分子吸附时Au(111)表面的吸附能最大, 而在解离吸附时AuCu(111)-Au具有最大的吸附能. 上述电子转移的计算结果说明了2.1节中不同吸附状态的能量关系.
图5 水分子在Au(111), Cu(111), AuCu(111)不同吸附状态沿表面法线方向的平均差分电子密度图
Fig.5 Profile of mean electron density difference of water on Au(111), Cu(111), AuCu(111) surfaces by the different adsorption states
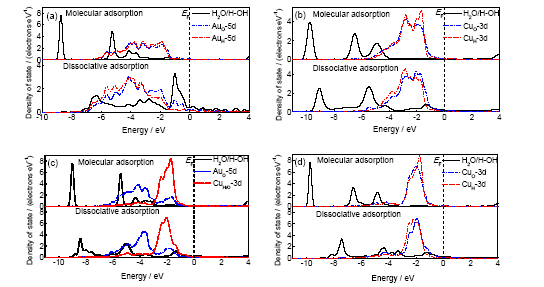
从电子结构的变化可以进一步说明水分子或者裂解产物在表面上吸附的异同, 以及它们之间的能量对比关系. 从图6可以看出, 在4种表面上的分子吸附时, 水分子的1b2电子态与孤立水分子是一致的, 水分子与表面的相互作用主要是通过其孤对电子态(包括3a1和1b1)与Au或Cu的d电子态相互杂化而发生作用. 但是在Au(111)表面和AuCu(111)-Au表面上水分子的3a1电子态与孤立水分子相比变化很小, 而在Cu(111)表面和AuCu(111)-Cu表面上时则变化较为明显. 同时水分子的1b1电子态在Au(111)表面和AuCu(111)-Au表面上完全与Au的d电子态重叠, 发生明显的杂化, 而在Cu(111)表面和AuCu(111)-Cu表面上时则变化较小. 另一方面, Cu的d电子态与Au的d电子态相比, 更容易形成靠近费米能级的孤立电子态. 这些计算结果说明, 对于分子吸附状态, Au与水分子的相互作用比Cu与水分子的相互作用更强; 对于解离吸附状态, H+OH基团的类1b2电子态在Au(111)表面上完全消失, 并且与3a1电子态一起与Au的d电子态完全重叠、杂化, 但是在Fermi能级EF附近有一个未完全杂化的类1b1电子态, 该电子态主要由顶位吸附的表面羟基形成, 与吸附位上的Au原子有一定的相互作用. 在其余3个表面上, H+OH基团表现出一定的类1b2和3a1电子态特征, 而其类1b1电子态则与吸附位上的d电子态完全重叠、杂化. 从另一方面来看, 由于Cu的d电子态比Au的d电子态更靠近Fermi能级, 根据金属催化材料的d带中心理论, 即Cu比Au具有更强的催化活性, 而且在有Cu存在的表面上, 水分子的1b1电子态或者H+OH基团的类1b1电子态都发生更为完全的重叠和更明显的杂化. 从这个角度可以解释在AuCu(111)表面上, 水分子裂解反应的反应能更小, 甚至在Cu(111)表面上解离吸附比分子吸附更稳定, 水分子的裂解反应成为放热反应. 这一变化的内在原因就在于水分子或H+OH基团与表面金属原子不同类型的相互作用, 以及金属原子d电子态所反映的活性不同.
图6 水分子在不同表面上以分子吸附或解离吸附时的局域分波态密度图
Fig.6 Local and partial density of states of water on Au(111) surface (a), Cu(111) surface (b), AuCu(111)-Au surface (c) and AuCu(111)-Cu surface (d) at the present of molecular adsorption or dissociation adsorption (EF—Fermi level)
(1) 在从分子吸附到解离吸附的反应路径上, 以反应活化能为比较标准, 水分子在所考虑的4种模型中的反应活性顺序如下: Au(111)<AuCu(111)-Cu<AuCu(111)-Au<Cu(111). 这与水分子的分子吸附状态在Au(111)表面和AuCu(111)-Cu表面具有相对较大的吸附能, 而解离吸附状态具有相对较小的吸附能有关. 与此相反的是, 水分子的分子吸附状态虽然在AuCu(111)-Au表面和Cu(111)表面具有相对较小的吸附能, 但同时解离吸附状态时的吸附能也相对较大.
(2) 从不同吸附状态与表面电子转移情况看, 两者之间电子转移较多, 则意味着相互作用较强, 对应的吸附能较大, 所以分子吸附时Au(111)表面的吸附能最大, 而解离吸附时AuCu(111)-Au的吸附能最大.
(3) 对于水分子在金属催化剂表面的裂解反应, 过渡金属Cu具有更高的活性, 因此在实际反应体系中, 用AuCu二元金属合金替代贵金属Au催化剂不仅可以降低材料成本, 还可以提高反应活性.
The authors have declared that no competing interests exist.

/
| 〈 |
|
〉 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}