
彭以超 , 杜晨阳
, 杜晨阳
PENG Yichao, DU Chenyang
中图分类号: TG113;TG132
文献标识码: A
文章编号: 0412-1961(2015)01-0011-10
通讯作者:
修回日期: 2014-05-21
网络出版日期: --
版权声明: 2015 《金属学报》编辑部 版权所有 2014, 金属学报编辑部。使用时,请务必标明出处。
基金资助:
作者简介:
彭以超, 男, 1989年生, 硕士生
展开
摘要
采用乙炔真空渗碳方式对服役6 a的乙烯裂解炉管合金Cr35Ni45Nb进行了加速渗碳处理, 并利用SEM, XRD及定量电子探针等手段对渗碳前后炉管内壁的渗碳行为及相演化机理进行系统研究. 结果表明, 炉管内表面形成了较厚的Cr2O3/SiO2复合氧化层. 复合氧化层表现出良好的抗腐蚀能力, 是阻止C渗入炉管内部的有效障碍. 材料的抗渗碳能力主要取决于表层Cr2O3的连续性、致密性和亚表层SiO2的稳定性. 在低氧分压且具有还原性的气氛中, 表层的Cr2O3层逐渐碳化为Cr3C2, 并且Cr3C2逐渐剥离和脱落, 使得保护性Cr2O3膜的抗渗碳能力逐渐减弱甚至消失. 亚表层的SiO2虽具有优良的热力学稳定性, 但Si较低的活度使得SiO2层不够连续, 仍有部分C从氧化层空隙间渗入. 当移除该复合氧化层或者渗碳时间足够长使得复合氧化层抗渗碳能力急剧减弱时, 炉管材料内部由于发生严重的内部渗碳使得组织结构发生了显著变化: 枝晶间碳化物严重合并和粗化, 并原位发生由M23C6到M7C3的转变, 同时在碳化物内部析出类似于离异共析状的蠕虫状g相. 距表面越近, C活度越高, 导致在约0.5 mm深的范围内发生以大量石墨析出为特征的金属尘化现象. 碳化物的严重合并粗化以及金属尘化现象会造成炉管组织的严重弱化、宏观裂纹的产生以及炉管服役寿命的降低.
关键词:
Abstract
Carburization in Ni-Cr-Fe-based alloys is an important phenomenon, especially in ethylene cracking tubes which serve at high temperatures under highly carburizing environment. In this work, the Cr35Ni45Nb tube subjected to service condition for 6 a was carburized by low-pressure vacuum carburizing (LPVC) at 1080 ℃. The carburization behaviors and corresponding mechanisms of phase evolution in the inner wall were comprehensively analyzed through SEM, XRD and EPMA. The results showed that oxidation behaviors of the tube at high temperature were consisted of external oxidation of Cr and internal oxidation of Si, resulting in formation of composite oxide scales. Depletion of Cr in the subsurface caused by surface Cr2O3 leaded to carbide dissolution and formation of carbide free zone and carburized zone. The critical concentration of Cr for carbide dissolution is about 19.0% (mass fraction). By comparing carburization behaviors of specimens whose oxide scales were retained or removed, the carburization resistance of the composite oxide scales in carburizing environment was systematically investigated. The results showed that the composite oxide scales formed previously acted as an effective barrier to carbon infiltration. However, the outermost Cr2O3 scale tended to be carbonized to form carbide scale to spall from the surface in the strongly reducing environment with low oxygen partial pressure, while the SiO2 kept stable all along due to its excellent thermodynamic stability. However, a certain amount of carbon was still capable to penetrate the alloy interior through gaps of the SiO2 scale due to its discontinuity. Therefore, continuity, density and high-temperature stability of the oxide scales were crucial for the alloy to achieve excellent anti-carburizing performance. Once the oxide layers were removed or carbonized adequately, inconceivable internal carburization occured widely. Large amounts of secondary carbides precipitated again in the previous carbide free zone due to high carbon activity. Widespread precipitations of graphite called metal dusting in the range of about 0.5 mm in depth occurred after long exposure of specimens to the carburizing environment. The carbon activity gradually decreased with increasing distance from the surface. The primary carbides within the deeper carburized region were transformed from M23C6 to M7C3 in situ, which were accompanied by precipitation of vermicular g phase in the primary carbides, phase transition from h to NbC and decomposition of intragranular secondary carbides. Severe coalescing and coarsening of carbides and metal dusting caused the serious degradation of microstructure, formation of macro-cracks and final thinning of the Cr35Ni45Nb tube wall.
Keywords:
乙烯裂解炉是石化工业生产乙烯的重要装置, 而辐射段炉管是裂解炉的关键部件, 辐射段炉管的任何失效形式都可能导致系统的非计划停车、爆炸或者更加严重的后果[1-3]. 服役过程中炉管内是高速的水蒸气和碳氢化合物组成的混合气体, 因此形成了强烈渗碳和轻微氧化的环境[4,5]. 虽然炉管内氧分压较低, 但是仍可以充分氧化Cr, Si等元素, 从而在炉管内壁形成一层氧化膜, 氧化膜具有一定的防氧化和抗渗碳作用[6-9]. 然而实际过程中仍然还有大量服役炉管由于受到严重渗碳腐蚀而发生故障, 这主要是由于温度变化、应力、蠕变、清焦等原因造成的氧化膜失效所导致[10-13]. 由于C的高扩散率, 渗碳会使碳化物聚合粗化, 使得合金内部组织形态和化学成分都发生较大的变化[14]. 在高C活度下, 严重的渗碳甚至会导致炉管发生灾难性的金属尘化[15-17]. 由氧化和渗碳造成的内壁组织的弱化使得炉管壁有效壁厚减薄, 持久寿命降低. 迄今, 有关炉管材料在高温长时服役条件下氧化行为的研究较多[18-20], 而有关炉管材料在高温服役过程中的渗碳及抗渗碳行为的研究相对较少.
本工作以服役6 a的Cr35Ni45Nb耐热合金炉管为研究对象, 通过高温真空低压加速渗碳实验对材料进行化学热处理, 系统地研究了该合金在接近高温服役条件下的渗碳行为, 包括渗碳动力学、复合氧化层的抗渗碳特性及其对渗碳行为的影响、渗碳后组织及相演变规律及更严重的金属尘化现象等, 旨在探索该合金在高温服役下的渗碳机理及氧化-碳化叠加机理, 从而为该合金炉管抗渗碳性能的提高、服役过程更换准则的建立以及进一步的服役寿命评估奠定基础.
实验材料为服役6 a (约52000 h)的Cr35Ni45Nb辐射段炉管, 其原始化学成分(质量分数, %)为: C 0.5, Nb 1.0, Cr 35.44, Ni 43.57, Ti 0.01, Si 1.6, Fe余量.
在服役炉管上分别切割用于组织分析的6块20 mm高环状炉管试样, 将其中一半试样的内壁打磨至恰好出现光亮表面, 而另一半维持原状. 另外, 为了研究合金的渗碳动力学, 在炉管内部切取18块20 mm×10 mm×5 mm的矩形块状试样, 此时各表面无氧化膜, 将其中一半的块状试样在SRJX-4-13箱式电阻炉中进行90 h预氧化实验从而得到表层氧化膜, 然后将所有矩形块状试样在丙酮中超声波清洗并烘干, 用Sartorius-BS223S电子天平称取初始质量. 将所有试样平均分为3批次分别放入双室真空渗碳炉中进行低压真空渗碳, 渗碳温度1080 ℃, 渗碳时间分别为1, 3和5 h. 渗碳结束后, 再将矩形试样称重, 从而获得渗碳的动力学曲线. 为了通过对比动力学曲线的不同趋势来评估合金抗渗碳能力的强弱, 也对未服役炉管和分别服役1.5和2.5 a的Cr35Ni45Nb炉管进行取样渗碳处理.
渗碳结束后, 对环形渗碳试样进行线切割、机械打磨、抛光及电解浸蚀, 利用DMAX-RB旋转阳极X射线衍射仪(XRD), JSM-6510A扫描电子显微镜(SEM), JED-2300型能谱仪(EDS)及JXA-8230电子探针系统(EPMA)分析渗碳后表面氧化层的组织结构变化和内部渗碳区域渗碳行为及相演变规律.
图1a为服役6 a的Cr35Ni45Nb耐热合金炉管内壁显微组织. 可以看出, 内壁主要分为3个典型区域: 表面的复合氧化层区、亚表层贫碳化物区和内部渗碳区. 图1b为表面复合氧化层区组织分布, 图1c~f分别为对应于图1b区域的Ni, O, Si, Cr的面分布, 表1中列举了图1b中各位置的EPMA定量分析结果. 结合能谱面分布和EPMA分析可以看出, 外表层的灰色相为Cr2O3, 其下方的黑色相为SiO2. SiO2下方为贫碳化物区, 整个氧化层厚度约80 μm. 从贫化区端点开始, SiO2仍然以晶间氧化的方式沿着贫化区晶界向内延伸一段距离, 平均深度约为62 μm, 贫化区宽度约230 μm, 晶间氧化区域宽度小于贫碳化物区域宽度. Bennett和Price[21]发现, 服役过程中的HK40合金的晶间氧化区的终点恰好为内部渗碳区的起点, 因而采用了一种“裂缝腐蚀”机制来解释晶间氧化及内部渗碳的机理, 而本研究结果显示, 晶间氧化区宽度要比贫碳化物区窄得多, 其原因是相对于HK40而言, Cr35Ni45Nb合金基体高含量的Cr保证了表面氧化层的致密性和连续性, 从而减少了环境中O和C的渗入, 使得晶间氧化区发展缓慢.
图1 服役态Cr35Ni45Nb炉管内侧横截面的组织特征及相应的元素面分布
Fig.1 Morphologies of inner cross section of Cr35Ni45Nb tube in service condition (a), surface composite oxide scales (b) and corresponding elemental mapping of Ni (c), O (d), Si (e), Cr (f)
表1 各图中所示位置的EPMA分析结果
| Position | C | Cr | Si | Ni | Fe | Nb | O | Phase |
|---|---|---|---|---|---|---|---|---|
| 1 | 5.88 | 32.97 | 0.03 | 0.26 | 0.74 | 0.08 | 60.04 | Cr2O3 |
| 2 | 9.19 | 0.17 | 25.14 | 0.16 | 0.13 | 0.00 | 65.22 | SiO2 |
| 3 | 38.75 | 41.93 | 0.27 | 9.58 | 9.37 | 0.10 | 0 | M3C2 |
| 4 | 0 | 0.72 | 27.17 | 0.94 | 0.79 | 0.06 | 70.31 | SiO2 |
| 5 | 33.89 | 52.30 | 0.03 | 2.86 | 10.77 | 0.14 | 0 | M7C3 |
| 6 | 78.75 | 2.05 | 0.13 | 1.61 | 1.12 | 0.03 | 16.31 | Graphite |
| 7 | 36.46 | 54.24 | 0.18 | 2.14 | 6.81 | 0.17 | 0 | M3C2 |
| 8 | 0 | 32.32 | 0.18 | 0.19 | 0.16 | 0.01 | 67.19 | Cr2O3 |
| 9 | 4.10 | 6.40 | 0.74 | 56.34 | 32.39 | 0.03 | 0 | g |
| 10 | 1.90 | 14.01 | 0.50 | 51.75 | 31.80 | 0.03 | 0 | g |
| 11 | 51.32 | 1.11 | 0.79 | 1.65 | 0.42 | 44.71 | 0 | NbC |
| 12 | 4.17 | 4.72 | 15.07 | 30.62 | 2.64 | 42.78 | 0 | h |
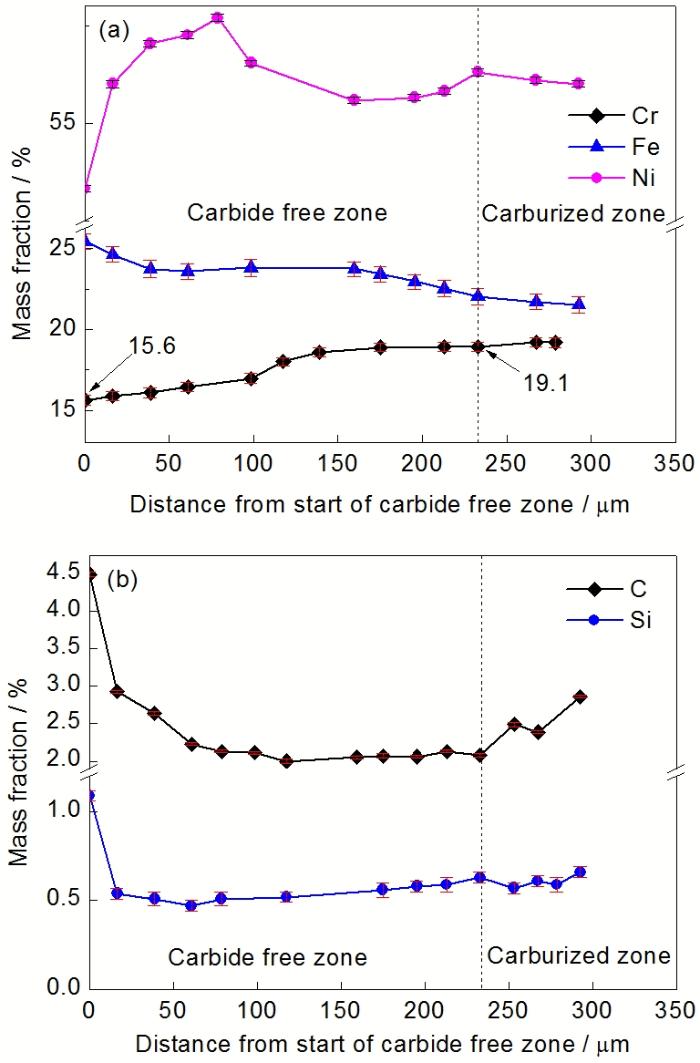
图2 服役6 a的Cr35Ni45Nb炉管贫碳化物区元素含量线分布
Fig.2 Concentration profiles of Cr, Ni, Fe (a) and Si, C (b) in carbide free zone of Cr35Ni45Nb tube serviced for 6 a
图2为服役6 a的Cr35Ni45Nb炉管贫碳化物区中各个元素的浓度(含测量误差)变化曲线. 由于每个测量点所选区域有限, 因此从理论上来说图2不能完整地代表整个贫化区元素浓素的线分布情况, 但从实际而言, 图2曲线的基本趋势以及关键位置的浓度已经基本可以反映实际情况, 仍具有较高的实用价值. 表面氧化层形成过程中, 由于亚表层基体中的Cr扩散到表层处与O形成氧化膜, 导致该区Cr含量降低(图2a)及形成碳化物的临界C浓度增高, 碳化物分解. 距氧化膜越近, Cr浓度越低, 表面氧化层附近Cr的浓度最低, 约15.6% (质量分数); 贫碳化物区的末端Cr含量约为19.0%, 说明19.0%为Cr35Ni45Nb合金贫碳化物区碳化物析出分解的临界浓度, Cr的浓度高于该值时碳化物可以稳定存在不发生分解, 并进一步导致内部碳化物富集区的形成. 当然, 该浓度对应的是低C活度下的临界浓度, 对于高C活度时碳化物的析出或分解并不适用. 从图2a亦可以看出, 贫碳化物区Ni的含量略有增加, 其原因在于Cr含量较内部基体相对降低所致. Cr含量的降低造成该区域的C活度升高, 使得向内的C分布趋势呈上坡扩散(图2b).
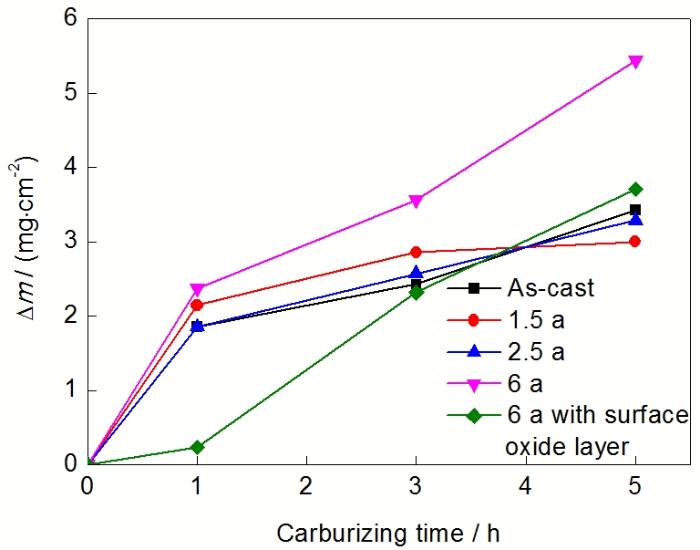
图3 1080 ℃下不同时间服役态Cr35Ni45Nb的真空渗碳动力学曲线
Fig.3 Vacuum-carburizing kinetic curves of Cr35Ni45Nb alloys with different service times at 1080 ℃(△m—mass gain)
2.2.1 渗碳动力学 图3所示为不同服役时间的Cr35Ni45Nb炉管在1080 ℃乙炔真空渗碳后的渗碳动力学曲线. 可以看出, 不同状态炉管材料的渗碳增重有较为相似的规律. 在无表面氧化膜的前提下, 服役时  间在0~2.5 a范围内, 各炉管的渗碳增重随时间变化曲线相差不大, 说明高镍铬材料在服役2.5 a后仍保持良好的抗渗碳性能. 而对于服役6 a的炉管, 无表面氧化层时渗碳增重明显高于其它服役态的炉管, 说明炉管服役6 a后自身抗渗碳性能已经大大减弱, 在服役过程中极易受到碳化侵蚀. 然而, 在表面预制氧化膜后再进行渗碳, 动力学曲线却发生了较大的偏差: 合金在初始1 h内渗碳增重极其缓慢地增长, 1 h后动力学曲线显著升高, 其斜率与无氧化膜时相近.
间在0~2.5 a范围内, 各炉管的渗碳增重随时间变化曲线相差不大, 说明高镍铬材料在服役2.5 a后仍保持良好的抗渗碳性能. 而对于服役6 a的炉管, 无表面氧化层时渗碳增重明显高于其它服役态的炉管, 说明炉管服役6 a后自身抗渗碳性能已经大大减弱, 在服役过程中极易受到碳化侵蚀. 然而, 在表面预制氧化膜后再进行渗碳, 动力学曲线却发生了较大的偏差: 合金在初始1 h内渗碳增重极其缓慢地增长, 1 h后动力学曲线显著升高, 其斜率与无氧化膜时相近.
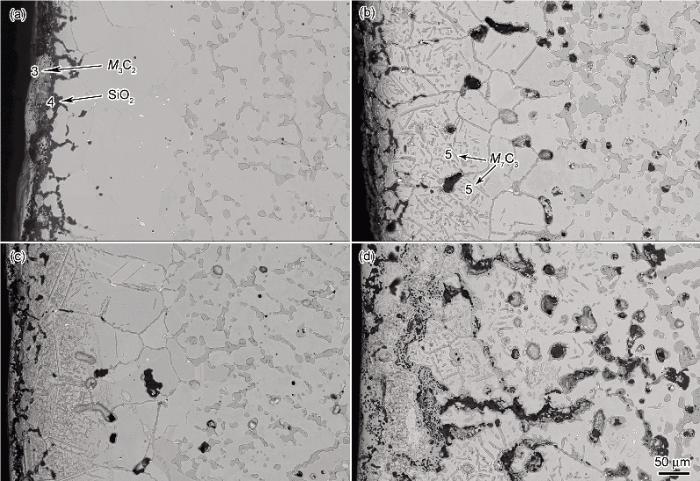
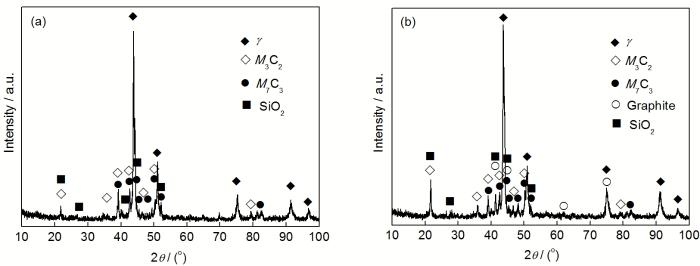
2.2.2 渗碳后炉管内侧组织特征 图4为服役6 a的Cr35Ni45Nb炉管真空渗碳后的内侧横截面显微形貌. 可以看出, 渗碳前后内壁组织发生了显著的变化. 对于未除去氧化膜的试样, 由于真空渗碳时表面C活度很高, 渗碳1 h后内壁的Cr2O3层就已逐步发生碳化, 结合图5a的XRD谱及表1的EPMA分析可知, 该碳化物为M3C2. 然而SiO2层在渗碳5 h后却依旧存在, 可知在1080 ℃真空渗碳环境下, 氧化区中的SiO2相对于Cr2O3非常稳定, 没有发生转化. 由于Cr2O3连续而且较厚, SiO2层较为致密, 孔隙、微通道较少, 并且C在氧化层中由于没有可测的溶解度, 使得复合氧化层具有优良的抗渗碳能力, 在真空渗碳1 h后合金内部几乎没有发生渗碳(图4a), 贫碳化物区依旧没有碳化物析出, 只有在局部区域由于裂纹的存在使得晶间氧化前沿的晶界析出较薄的碳化物.
图4b为未除去氧化层的炉管内壁渗碳5 h后的组织. 可以看出, 表面的Cr2O3层基本已经全部原位碳化, 失去了抗渗碳能力. 碳化层由于体积膨胀, 并且其与SiO2层或与合金基体的结合能力减弱, 使得图4b中的表面碳化层大量脱落. 在持续高温渗碳环境下, C可以沿SiO2中的没有完全合并的空隙向内部快速渗入, 贫化区晶界生成了较多碳化物. 由于通过体扩散的C含量较少, 因而晶内析出碳化物形态为横跨晶粒、类似孪晶界的平行片层碳化物和呈一定夹角的针状碳化物, 与奥氏体基体呈一定取向关系. 结合图5b的XRD谱和表1的EPMA分析可知该碳化物为M7C3. 此外, 与图4a对比可知, 图4b中由于石墨化析出引起的内部黑色斑块也慢慢增多. 从图5中XRD谱也可以看出, 由于真空渗碳1 h后石墨化析出很少, 因而图5a中的石墨峰还非常微弱, 基本掩盖在背底中, 而随着渗碳时间延长至5 h, 石墨化析出逐渐增多, 使得图5b中石墨峰的强度逐渐增强.
图4 不同条件下服役6 a的Cr35Ni45Nb炉管真空渗碳后内壁横截面显微组织形貌
Fig.4 Inner cross-sectional microstructures of Cr35Ni45Nb tube after 6 a service under different low-pressure vacuum carburizing (LPVC) times
图5 表面存在氧化膜的Cr35Ni45Nb炉管内壁真空渗碳不同时间后的XRD谱
Fig.5 XRD spectra of inner carburized walls of Cr35Ni45Nb tube containing oxide scale with LPVC for 1 h (a) and 5 h (b)
图4c是将内壁Cr2O3及部分SiO2氧化膜除去后渗碳1 h的组织. 由于有效的氧化层障碍减少, C可以通过晶界扩散和体扩散向内大量渗入, 渗碳深度约220 μm, 碳化物组织典型形貌特点是类孪晶碳化物和颗粒碳化物群. 由于晶内存在较大体扩散C通量, 因而相比于图4b而言, 晶内析出的碳化物群颗粒细小而均匀.
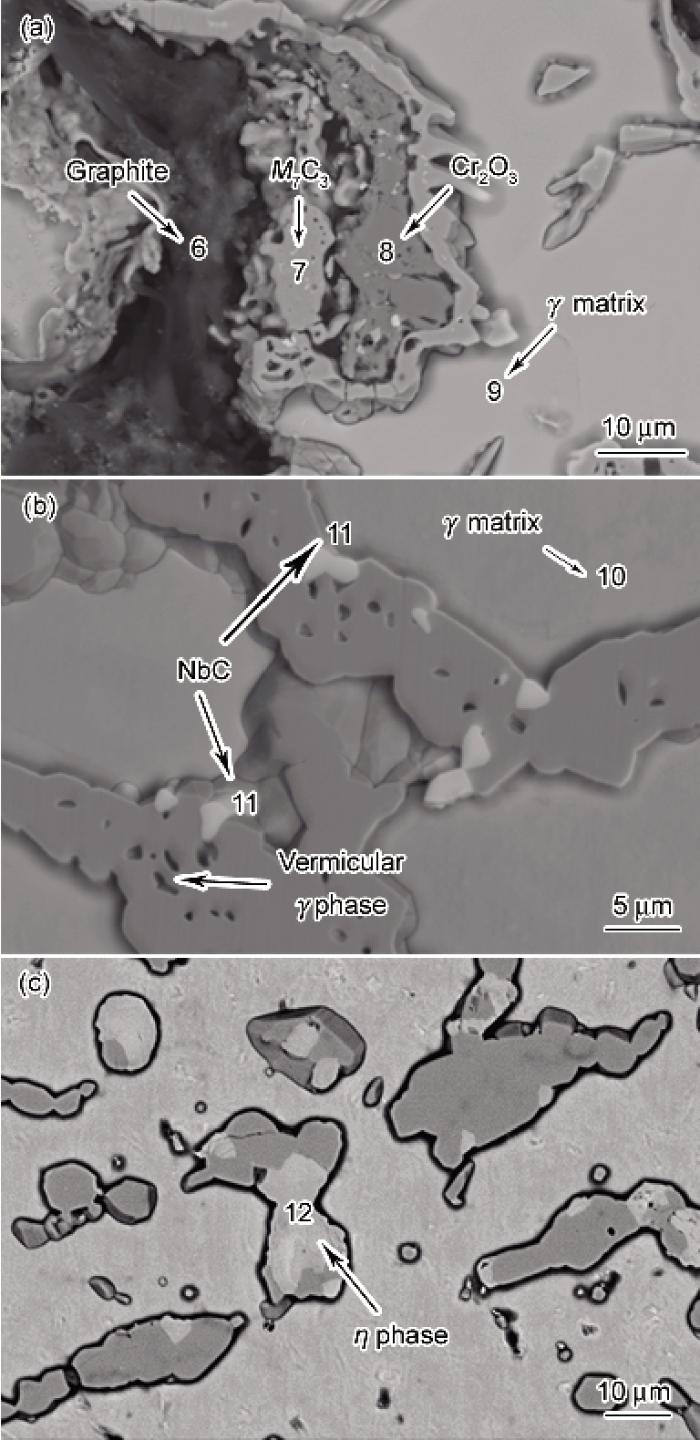
图4d是将内壁氧化膜除去后渗碳5 h的组织. 由于缺乏氧化膜的抗渗碳作用, 且渗碳时间较长, 因而合金内部出现了非常严重的渗碳, 在深度约510 μm的范围内普遍存在石墨化析出的现象. 图6是由于炉管内部石墨化析出导致的不同渗层深度的组织形态. 对图6a所示的石墨化区域不同位置进行EPMA分析, 结果如表1所示. 可知, 图6a中位置6对应的黑色区域主要为含夹杂的石墨, 7为M3C2, 8为原内氧化区域形成的Cr2O3, 9为g基体. 利用Thermo-Calc软件计算得到g基体的固溶度曲线如图7所示. 由表1中位置9的成分可知, 该处的Cr原子分数为6.40%, 质量分数为6.39%, 根据图7可推知该处g基体的饱和C浓度为0.95%, 而实际测得位置9的C原子分数为4.10%, 质量分数为0.947%, 从而可知, 碳化物周围g基体中的C浓度基本已经饱和. 石墨化区域下方一段范围内渗碳后一次碳化物M23C6逐渐转化为M7C3, 并形成了蠕虫状g相, 如图6b所示. 图6b和c中的位置10为g基体, 11为NbC, 12为h相. 从而可知, 虽然原始态中的NbC经过6 a的长期服役缓慢演变为h相(Nb3Ni2Si)[22], 在高温1080 ℃及较高C活度下, 严重渗碳区域中的h相又重新转变成白色分散颗粒的NbC相, 而在炉管中心Nb依旧以块状的h相存在(图6c). 石墨化区域下方g基体内却并无二次碳化物析出, 对图6b的位置10进行EPMA分析(表1)可以知道, 该区域C活度已经明显下降, g基体的Cr原子分数为14.01%, 质量分数为13.58%, 结合前面贫碳化物区形成原理的分析可知, 该区域Cr含量低于碳化物分解的临界质量浓度19.0%, 二次碳化物皆不能稳定存在而不析出, 从而出现枝晶间碳化物转变粗化、晶内贫碳化物的独特组织特点.
图6 移除表面氧化膜的服役6 a的Cr35Ni45Nb炉管真空渗碳5 h后的内部组织
Fig.6 Inner microstructures of Cr35Ni45Nb tube after 6 a service removing oxide scale with LPVC for 5 h(a) graphitization area(b) beneath the graphitization area(c) center of the tube
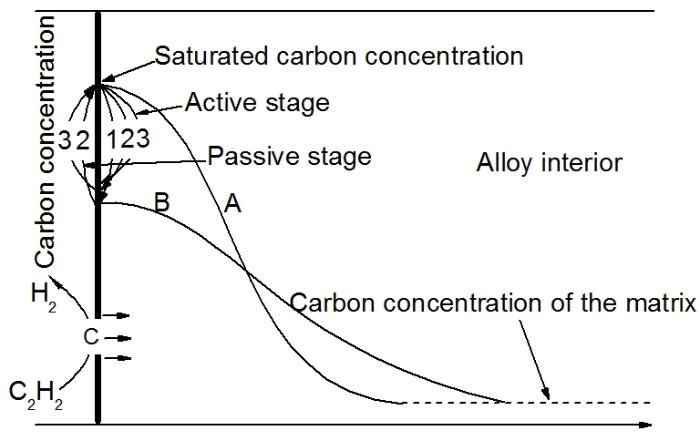
高温渗碳过程涉及3个独立的动力学过程[23]: (1) C从乙炔气体传递到炉管表面, 形成含碳复合体; (2) C从合金表面扩散至合金内部; (3) C在合金内部与碳化物形成元素Cr, Fe结合形成碳化物. 炉管在高温环境下乙炔真空低压渗碳过程具有一定的特点. 图8为真空渗碳过程中渗碳期和扩散期结束时内壁C浓度分布曲线. 由于渗碳工艺中渗碳期(active stage)与扩散期(passive stage)交替循环的周期性, 使得C呈“循环波浪式”向合金内部渗入. 如图所示, 表面C浓度在渗碳期达到最高, 形成薄层碳化物, 在扩散期由于乙炔停止供给, 表层形成的部分碳化物颗粒又逐渐溶解并向内部扩散, 使得表面C浓度降低, 碳化物分布曲线变得平缓, 再次进入渗碳期后表面又重新达到饱和. 如此循环往复, 使得最终渗碳区域得以形成.
图8 真空渗碳过程中不同阶段结束时内壁C浓度分布曲线
Fig.8 Carbon concentration profile inside of furnace tube at the end of different stages of vacuum carburizing (A—active stage, B—passive stage)
乙炔在Fe, Ni的催化下会发生分解:
试样渗碳后单位面积上的渗碳增重
(
式中, A为试样的总表面积, DC为C在合金中的扩散系数, k为反应速率常数, gC为C活度系数,
即渗碳速率和时间呈抛物线关系, 与图3中采用无氧化膜的矩形试样测定的动力学曲线规律一致. 若表面存在氧化层, 则表面反应速率成为主要控制因素, 即
图3中采用表面存在氧化膜的矩形试样测定渗碳动力学曲线时, 该预制的氧化膜主要成分为Cr2O3. 由于Si含量较低且预氧化时间相对较短, SiO2仍为分散的颗粒, 此时其抗渗碳能力可忽略不计. 结合图3中含氧化膜的动力学曲线分析可知, 在初始1 h内, 由于氧化膜的抗渗碳作用, C与氧化膜的表面反应成为影响渗碳速率的主导因素, 使得在同样的
C在Cr2O3氧化层并无可测的溶解度, 因而C无法穿过致密的氧化膜. 但服役过程中氧化膜并不能一直保持连续致密的状态, 氧化层中的孔隙和微通道的形成机制为: (1) 氧化层的分解, 特别在晶界上, 在分解压很低时, 适用于低压扩散阶段; (2) 在氧化层加厚过程中由于生长应力导致的破裂, 适用于当氧化层较厚的时候; (3) 生长晶粒的不完美交接, 适用于本研究中的Cr2O3层. 并且, 由于服役过程中高温蠕变过程造成的氧化层张力、氧化层与基体之间结合产生的张力、清焦过程中的热冲击以及温差应力等, 都有可能使表层氧化膜内部出现各种缺陷, 贯穿氧化层的缺陷可以作为含C分子的扩散途径, 从而使得C可以通过这些氧化层缺陷渗入合金内部.
实际上, 本研究中氧化层中的渗碳不止上述从缺陷渗入一种机制, 在高温、低氧分压且具有还原性的乙炔气氛中, 表面Cr2O3氧化层本身并不稳定, 在高C活度下逐渐发生碳化, 导致本身抗渗碳能力的逐渐退化. 在渗碳区域只发现了Cr3C2和Cr7C3的存在(图5和表1), 氧化物的碳化过程可用下式表示:
在高碳势下, Cr3C2的稳定性高于Cr7C3和Cr23C6, 因而作为第一反应产物形成. 若在氧化层内随反应深度的增加, 氧化层内部由于碳势降低也会发生如下反应:
即Cr2O3被碳化成Cr7C3. 或者已生成的Cr3C2与底部的Cr2O3发生作用:
也会有Cr7C3的生成, 但仍以M3C2为主要的反应产物.
事实上, 上述沿缺陷扩散和氧化层碳化这2个机制也是相互促进的, 大量的氧化层孔隙和微通道使得含碳气氛能够深入氧化层内部发生分解和与氧化物颗粒发生反应, 从而加速了氧化层的还原, 而生成的碳化物颗粒也会使得氧化层内部缺陷进一步增多和扩张.
图9为炉管表面氧化层发生碳化后碳化物层剥落的表面及侧面形态. 可见, 当渗碳时间较长, 使得表面大部分区域Cr2O3氧化层皆变为碳化物层时, 碳化层与亚表层的结合状态相对于原先发生了较大变化, 且碳化层本身的热膨胀系数低, 碳化物层不足以承受原氧化层中的张应力, 造成了碳化物层的大量脱落. 当Cr2O3完全碳化后, 其本身的抗渗碳能力逐渐降至最低. 值得注意的是, 本研究中采用的是高温高碳势环境的加速渗碳实验, 实际服役过程中温度和碳势都要低很多, 因而氧化物碳化的动力学过程会大大延长.
相对于外表层的Cr2O3, 亚表层的SiO2则表现出了优异的稳定性, 渗碳5 h后仍几乎保持不变, 从而为炉管壁的抗渗碳能力提供了一定的保证. SiO2没有发生还原的原因在于SiC的热力学稳定性远远不如Cr3C2和Cr7C3, 因而在氧分压极低(可认为接近于0)和高还原性的C2H2气氛中仍可以保持不变, 因而不具备SiC形成的条件, 推测可知温度是影响其高温稳定性的决定因素. 由于SiO2致密且C溶解度极低, 使得其本身具有出色的抗渗碳能力. 但由于合金中Si含量低于形成连续SiO2氧化膜的临界浓度, 根据Wagner理论[25], SiO2颗粒不能横向生长彼此连接起来, 因而SiO2氧化层并不连续, C可以通过氧化层的间隙渗入合金内部, 从而抗渗碳性能减弱.
图9 服役态Cr35Ni45Nb炉管表面碳化后碳化物层剥落的表面及侧面形态
Fig.9 Surface (a) and cross-sectional (b) morphologies of Cr35Ni45Nb tube in service condition illustrating spallation of carbide scale after carburization
综上可知, 复合氧化层的抗渗碳能力是Cr2O3层和SiO2层协调作用的结果, 从而可以最大限度地阻止C的渗入. 氧化层的连续性、致密性和高温稳定性是决定氧化层抗渗碳能力的3个重要因素, 缺一不可. 因而本研究对于实际应用的炉管抗渗碳性能的启发是: (1) 在不降低主要力学性能的前提下, 合理调节合金中Si, Al等元素的含量, 使得表面复合氧化层具有良好的连续、致密及高温稳定性; (2) 清焦过程中伴随的复合氧化层的破坏和重建要保证氧化层在裂解气通入之前氧化膜能够完全恢复至连续、致密的最佳状态.
服役6 a炉管除去表面氧化膜后, 抗渗碳能力大大减弱, 由图3显示, 在同样渗碳环境下渗碳5 h后的渗碳增重为4.714 mg/cm2, 比服役1.5 a的炉管增长57.1%. 服役6 a炉管渗碳前一次碳化物已经严重合并和粗化, 宽度在4~9 μm之间; 渗碳后碳化物宽度增至20~40 μm, 最大处甚至达80 μm宽左右. 由于碳化物宽度迅速扩展, 使得碳化物周围短距离内基体中的Cr向碳化物迅速扩散, 基体中Cr含量急剧下降, 造成严重敏化态晶间腐蚀. 在无石墨化渗碳区域, 碳化物转变可以用下式来表示:
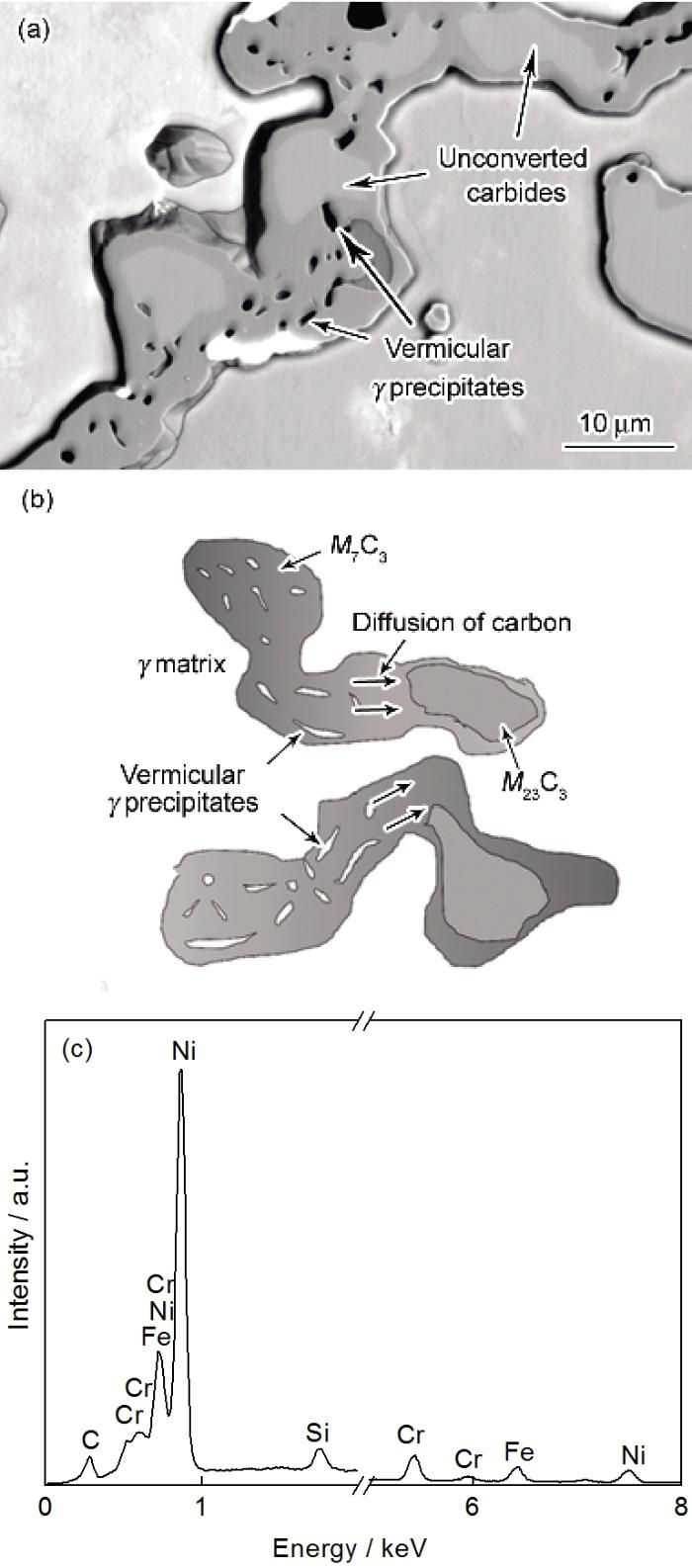
服役炉管材料在未发生渗碳时, C活度很低, 炉管一次枝晶间碳化物以M23C6的形式稳定存在; 而发生渗碳后, 渗碳区域C活度升高, 一次碳化物逐渐发生M23C6到M7C3的转变. 图10a和b分别为该碳化物转变的形态及示意图, 从形态上看, 这种转变类似于离异共晶, 但由于是固-固转变, 因而可认为是一种离异共析结构. 该碳化物转变会导致原来固溶于M23C6的部分原子发生出溶, 一部分进入周围g基体, 另一部分因为动力学因素无法迅速体扩散至碳化物-g基体界面而在碳化物内部形成蠕虫状g相. 图10c为蠕虫状g相的EDS谱, 一次碳化物中大量的晶格缺陷为蠕虫状g相的形成提供了形核位置. M7C3中蠕虫状g相的形态会随着距表面距离的减少(即C活度的升高)而发生粗化、延长和相互融合, 这种现象有利于服役过程中裂纹在枝晶干内的扩展.
图10 高C活度下M23C6向M7C3的转变形态, 转变示意图及蠕虫状g相的EDS谱
Fig.10 Morphology of carbide transition from M23C6 to M7C3 at high carbon activity (a), schematic of carbide transition (b) and EDS analysis of vermicular g precipitates (c)
随着距表面距离的减少C活度增大, 粗化的一次碳化物内部蠕虫状g相互连接, 并产生大量缝隙, 持续的渗碳气氛可沿着蠕虫状g相中的通道进入合金内部, 剩余的超饱和的C以石墨的形式在蠕虫状g相中沉积出来, 这是一种金属尘化现象, 真空渗碳环境的高C活度和低氧分压为金属尘化的发生提供了条件.
关于服役6 a炉管去除氧化层渗碳后发生严重金属尘化的机理, 有以下2种推测: (1) 一次碳化物周围基体中的Cr含量已经降至约6.4%左右, 不足以继续形成大量的碳化铬, 因而大量乙炔在一次碳化物内部蠕虫状g相中Fe, Ni的催化下形成大量石墨; (2) 乙炔在金属的催化作用下分解, 形成的C会立即被金属基体吸收, 使得金属中的C趋于饱和, 生成稳定碳化物和不稳定碳化物, 不稳定碳化物在一定条件下发生分解形成金属颗粒和C, 从而导致金属碎化和石墨沉积. 由于石墨和碳化物的密度和基体差别较大, 晶界大量石墨和碳化物析出和严重粗化, 使得整个晶粒发生撬起, 产生宏观裂纹(图11), 最终导致炉管壁的减薄和金属尘化的继续深入. 当然, 发生金属尘化的前提是炉管本身在6 a服役的过程中, 由于晶间氧化或内部渗碳作用使得炉管壁附近基体中的Cr被大量消耗, 抗渗碳能力减弱, 容易晶间腐蚀. 表面的氧化层对于延缓或抑制金属尘化的发生具有良好的效果. 若炉管渗碳前未去除氧化层, 发生金属尘化则需要经历一个孕育期, 即发生氧化膜的碳化、破裂和内碳化物的形成.
在炉管内部非渗碳区域中, 原始铸态管中的共晶NbC在长期高温时效过程中, 会逐渐吸收Ni, Si原子和排出C原子, 从而逐渐转化为化学计量为Nb3Ni2Si的h相(图6c). 事实上, NbC在1080 ℃下较为稳定, 然而由于服役过程中乙烯裂解大量吸热, 使得炉管内部大部分区域温度大幅下降, 温差较大, 内外壁之差甚至可达200 ℃左右, 因而实际服役温度要小于标准设定的1080 ℃. 这样的温差使得大量的NbC变为不稳定状态, 从而加速向铌镍硅化物转变, 所以温度是影响h相是否稳定存在的重要因素.
图11 金属尘化产生的宏观裂纹形貌
Fig.11 Macroscopic morphology of crack resulted from metal dusting
1080 ℃真空渗碳后, 在炉管内部渗碳区域, h相又重新转化为NbC, 这意味着在1080 ℃且较高碳活度下, h相是不稳定的, 温度是促进这一转化的前提, 高C活度则是加速这一转化的重要条件, 原因是由于Nb是强碳化物形成元素, 高C活度打破了NbC
(1) 当存在复合氧化层时, 氧化层是阻止C渗入的有效障碍, 氧化膜的抗渗碳能力决定于其连续性、致密性和高温稳定性. 复合氧化层的抗渗碳能力是由于Cr2O3层良好的连续性和致密性和厚度以及SiO2层良好的致密性和稳定性, 使得2者之间协调、配合从而最大限度地阻止C的渗入.
(2) 在低氧分压且具有还原性的乙炔气氛中, 表层Cr2O3逐渐碳化为Cr3C2, Cr3C2逐渐剥离和脱落, 使得Cr2O3的抗渗碳效果逐渐减弱至消失. 亚表层SiO2由于优良的热力学稳定性因而不发生变化, 但由于Si的活度较低使得SiO2层不够连续, 仍有部分C从氧化层空隙间渗入.
(3) 当移除该复合氧化层或者渗碳时间足够长使得复合氧化层抗渗碳能力急剧减弱时, 炉管材料内部的组织结构发生巨大变化: 枝晶间碳化物严重合并和粗化, 并原位发生由M23C6到M7C3的转变, 同时在碳化物内部析出类似于离异共析状的蠕虫状g相; 在高C活度及1080 ℃下, 由长期演化形成的h相又重新还原为NbC.
(4) 由于碳化物周围g基体的严重贫Cr和富C, 造成碳化物中的碳过饱和; 距表面越近, C活度越高, 导致在约0.5 mm深的范围内发生以大量石墨析出为特征的金属尘化现象; 碳化物的严重合并粗化以及金属尘化现象会造成炉管组织的严重弱化、宏观裂纹的产生以及炉管壁的减薄.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|

/
| 〈 |
|
〉 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}