
文怀梁 , 柯伟
, 柯伟
WEN Huailiang, KE Wei
中图分类号: TF777.1
通讯作者:
收稿日期: 2013-08-18
修回日期: 2013-12-5
网络出版日期: --
版权声明: 2014 《金属学报》编辑部 版权所有 2014, 金属学报编辑部。使用时,请务必标明出处。
基金资助:
作者简介:
文怀梁, 男, 1988年生, 硕士生
展开
摘要
在除氧的
关键词:
Abstract
As a kind of clean, efficient and relatively safe energy, nuclear energy has been widely used around the world. The high-level radioactive waste (HLRW) generated in the nuclear has also become a major risk, so the disposal safety of HLRW will be especially important. The planned concept of China's HLRW disposal program is a shaft-tunnel model located in saturated zones in granite. The metal container for sealing the HLRW is the key because its interaction with the ground water will lead to the leak of the HLRW during the long repository time. Beishan is a selected repository area and the ground water contains a bicarbonate (
Keywords:
作为一种清洁、高效和相对安全的能源, 核能已被世界各国普遍采用. 然而, 作为核能发电重要成本之一的高放废物安全处置问题也日益凸显. 我国高放废物处置拟采用深地质处置的方案, 即将玻璃固化后的高放废物放入密封的金属罐体中, 而后深埋在数百米的地下几千年甚至上万年[
实验用NaHCO3电解质溶液浓度依次为0.01, 0.02, 0.05和0.1 mol/L, pH值为8.33, 采用分析纯试剂和蒸馏水配制. 环境温度用水浴控制为30 ℃.
研究电极材料为Q235低碳钢, 化学成分(质量分数, %)为: C 0.18, Si 0.25, Mn 0.5, S 0.018, P 0.016, Cu 0.01, Cr 0.01, Ni 0.01, Al 0.02, Fe余量. 将其切割成10 mm×10 mm×3 mm的块状试样, 工作面积100 mm2, 其余表面用环氧树脂绝缘封装. 参比电极为饱和甘汞电极(SCE), 辅助电极为Pt片. 工作电极用砂纸逐级打磨, 然后用蒸馏水清洗, 乙醇除油, 干燥备用.
实验前, 预先向盛有电解质溶液的密封电解池中通入高纯N2约30 min, 以除去其中的溶解O2, 并始终在电解池的空隙处充满高纯N2以隔离空气接触. 用M273A型电化学工作站进行动电位极化曲线和电化学阻抗谱(EIS)的测量, 所有测量电位均相对于饱和甘汞电极的电位. 测量动电位极化曲线时, 扫描速度为10 mV/min, 从相对于开路电位-250 mV向正方向扫描. EIS测量的正弦波扰动电压幅值为10 mV, 测量的频率范围为10-2~105 Hz. 将低碳钢在4种不同浓度HCO3-溶液中的阻抗谱用ZSimpWin软件进行拟合, 并用等效电路模型进行分析. 采用HA-151A恒电位仪对低碳钢电极进行原位的腐蚀电位监测. 开路电位监测结束以后, 采用XD-5A型X射线衍射仪(XRD)对低碳钢电极表面生成的腐蚀产物进行分析. 而后用加有缓蚀剂的酸洗液(20 g (CH2)6N4+500 mL HCl+500 mL H2O)将电极表面的腐蚀产物除去, 用S-3400N型扫描电子显微镜 (SEM)观察其表面腐蚀形貌.
图1所示为裸低碳钢电极在不同浓度
极化曲线阴极支表明, 阴极电流密度随
图 1. 低碳钢在除氧
Figure 1. Polarization curves of low carbon steel in 0.01 mol/L(a), 0.02 mol/L (b), 0.05 mol/L (c) and 0.1 mol/L (d)deaerated
低碳钢电极在不同环境中的阳极极化曲线随
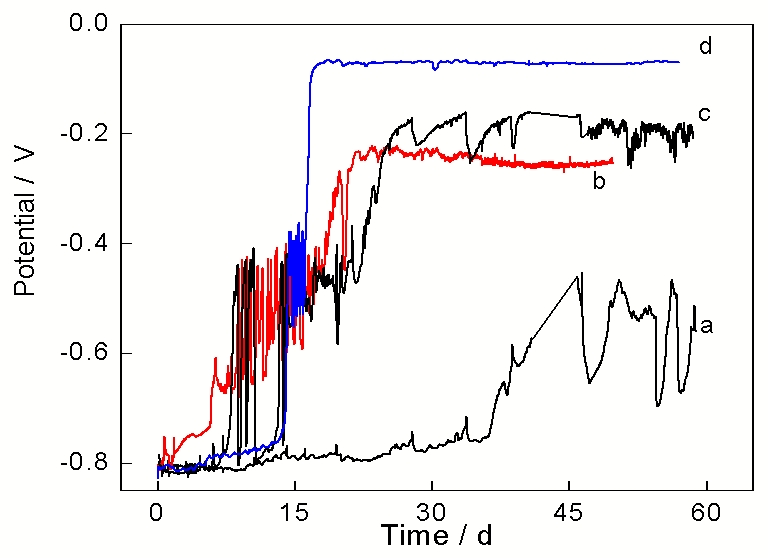
图2所示为低碳钢电极在4种不同
图 2. 低碳钢在除氧
Figure 2. Evolution of open circuit potential in 0.01 mol/L(a), 0.02 mol/L (b), 0.05 mol/L (c) and 0.1 mol/L (d)deaerated
在0.02 mol/L
在0.05 mol/L
在0.1 mol/L
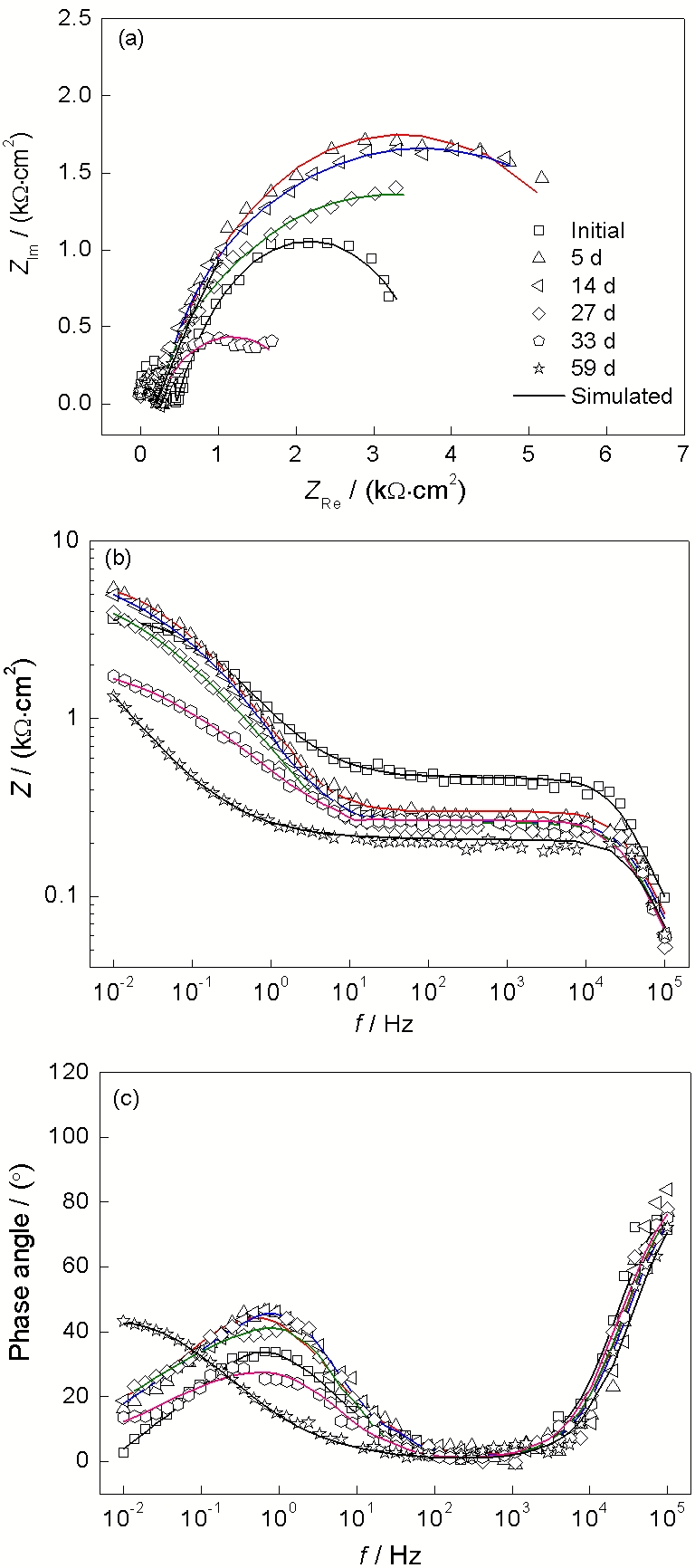
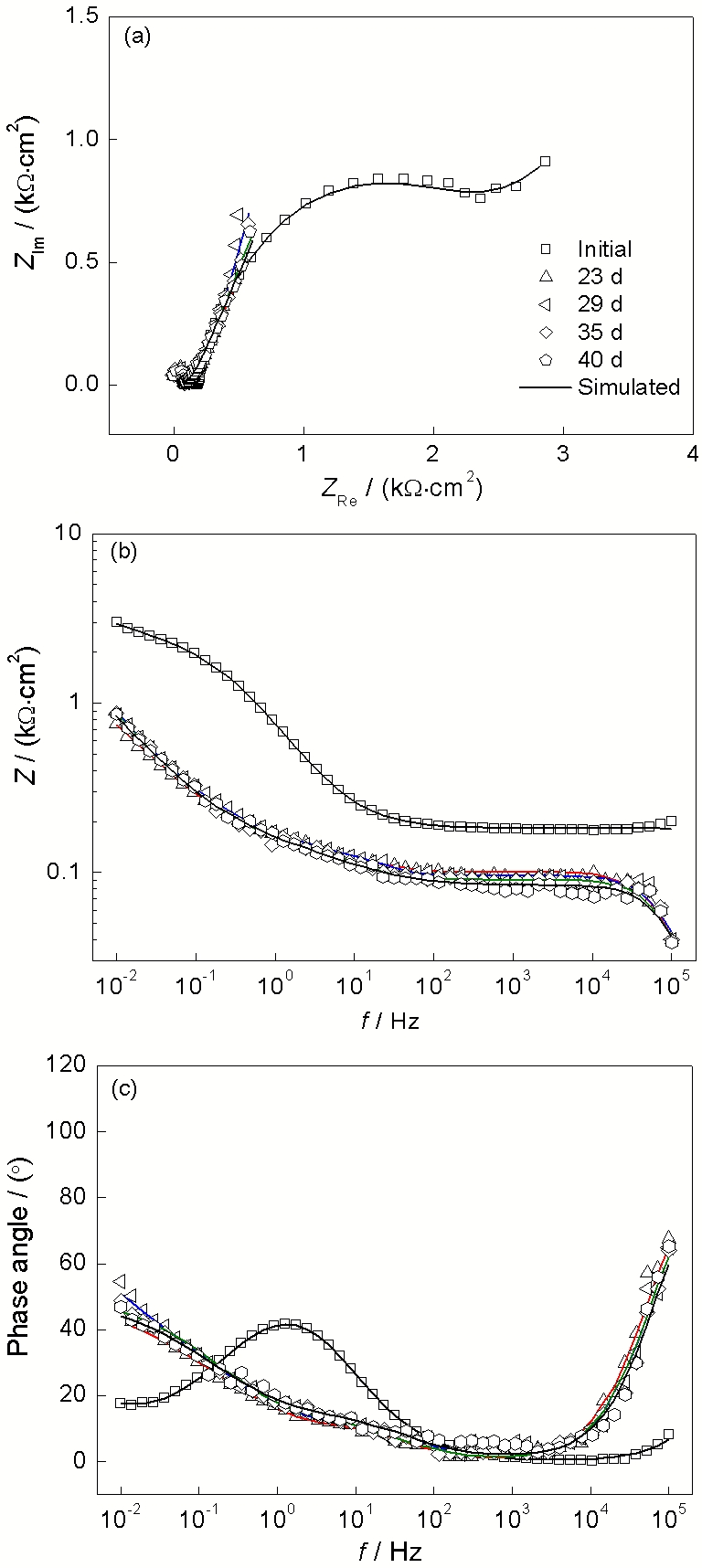
图3~6所示为低碳钢电极在4种溶液中长期浸泡的阻抗谱曲线, 以代表低碳钢表面电化学状态的演化特征. 其中图3为低碳钢在0.01 mol/L
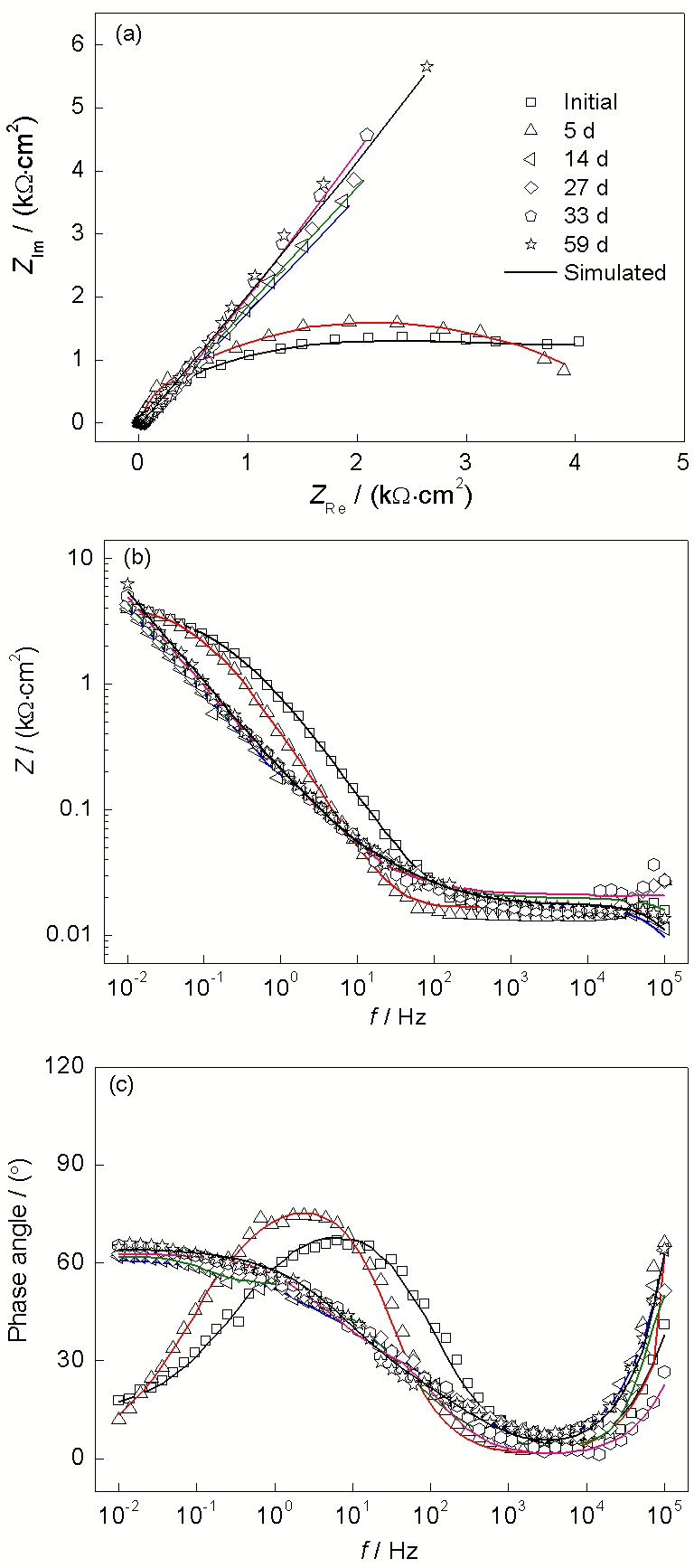
在0.02, 0.05和0.1 mol/L
图 3. 低碳钢在含有0.01 mol/L
Figure 3. Measured EIS results of low carbon steel in 0.01 mol/L
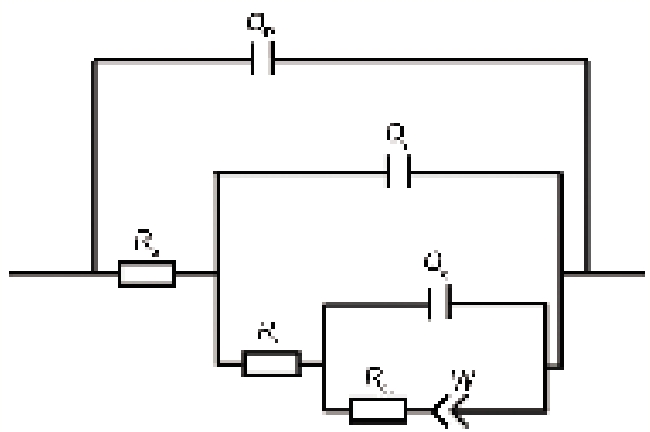
将低碳钢在4种不同浓度
式中, Y0, j, n均是常数, Y0量纲为S·sn·cm-2, j是虚数单位
表1~4所示为由图7的等效电路拟合得到的各元件参数. 拟合结果表明, 在长期浸泡过程中, 锈层电阻Rr随浸泡时间延长而增大, 这是由腐蚀产物随浸泡过程的进行而增多, 腐蚀产物层增厚造成的. nr在0和1之间, 说明锈层不均匀或者存在缺陷[
图 4. 低碳钢在含有0.02 mol/L
Figure 4. Measured EIS results of low carbon steel in 0.02 mol/L
图 5. 低碳钢在含有0.05 mol/L
Figure 5. Measured EIS results of low carbon steel in 0.05 mol/L
图 6. 低碳钢在含有0.1 mol/L
Figure 6. Measured EIS results of low carbon steel in 0.1 mol/L
图 7. 拟合电化学阻抗谱曲线的等效电路
Figure 7. Equivalent circuit for fitting the EIS data (Qp—capacitance caused by high frequency phse shift, Rs—solution resistance, Qr—rust capacitance, Rr—real part of electrochemical impedance, Qdl—double layer capacitance, Rct—charge transfer resistance, W—Warburg impedance)
同时, 拟合结果表明在4种溶液中, 扩散阻抗参数的数量级分别为102, 100, 10-4, 10-17. 即Yw随
表1 低碳钢在0.01 mol/L
Table 1 Fitting results for EIS plots in 0.01 mol/L
| Time / d | Yr mS·sn·cm-2 | nr | Rr Ω· cm2 | Ydl mS·sn·cm-2 | ndl | Rct kΩ·cm2 | Yw mS·s0.5·cm-2 |
|---|---|---|---|---|---|---|---|
| Initial | - | - | - | 0.33 | 0.7 | 3.4 | - |
| 5 | 0.28 | 0.5 | 2.7×10-5 | 0.11 | 1.0 | 6.7 | - |
| 14 | 0.11 | 1.0 | 15 | 0.33 | 0.5 | 6.9 | - |
| 27 | 0.14 | 1.0 | 46 | 0.52 | 0.5 | 6.6 | - |
| 33 | 0.66 | 0.8 | 62 | 0.83 | 0.2 | 6.9 | - |
| 59 | 1.40 | 0.9 | 89 | 3.60 | 0.6 | 7.1 | 120 |
表2 低碳钢在0.02 mol/L
Table 2 Fitting results for EIS plots in 0.02 mol/L
| Time / d | Yr mS·sn·cm-2 | nr | Rr Ω·cm2 | Ydl mS·sn·cm-2 | ndl | Rct kΩ·cm2 | Yw mS·s0.5·cm-2 |
|---|---|---|---|---|---|---|---|
| Initial | - | - | - | 0.34 | 0.7 | 2.2 | - |
| 23 | 0.21 | 1.0 | 26 | 6.00 | 0.5 | 1.0 | 0.60 |
| 29 | 0.13 | 1.0 | 31 | 5.50 | 0.6 | 1.6 | 0.32 |
| 35 | 0.27 | 1.0 | 38 | 5.00 | 0.6 | 1.9 | 1.30 |
| 39 | 1.10 | 0.7 | 71 | 5.50 | 0.6 | 1.9 | 4.0×10-3 |
表3 低碳钢在0.05 mol/L
Table 3 Fitting results for EIS plots in 0.05 mol/L
| Time / d | Yr mS·sn·cm-2 | nr | Rr Ω·cm2 | Ydl mS·sn·cm-2 | ndl | Rct kΩ·cm2 | Yw mS·s0.5·cm-2 |
|---|---|---|---|---|---|---|---|
| Initial | - | - | - | 0.35 | 0.7 | 2.4 | - |
| 5 | 0.12 | 0.9 | 0.06 | 0.28 | 0.6 | 2.6 | - |
| 23 | 0.43 | 0.8 | 5.90 | 0.60 | 0.6 | 2.8 | 5.6×10-5 |
| 39 | 0.35 | 0.7 | 22.0 | 0.19 | 0.6 | 2.4 | 2.1×10-4 |
| 46 | 0.07 | 1.0 | 34.0 | 0.47 | 0.6 | 1.9 | 1.7×10-4 |
| 52 | 0.07 | 1.0 | 48.0 | 0.72 | 0.7 | 1.5 | 3.4×10-4 |
表4 低碳钢在0.1 mol/L
Table 4 Fitting results for EIS plots in 0.1 mol/L
| Time / d | Yr mS·sn·cm-2 | nr | Rr Ω·cm2 | Ydl mS·sn·cm-2 | ndl | Rct kΩ·cm2 | Yw mS·s0.5·cm-2 |
|---|---|---|---|---|---|---|---|
| Initial | - | - | - | 0.29 | 0.8 | 3.90 | - |
| 9 | 0.16 | 0.5 | 1 | 0.34 | 1.0 | 4.70 | - |
| 23 | 0.07 | 1.0 | 11 | 1.40 | 0.6 | 2.00 | 4.9×10-7 |
| 26 | 0.13 | 1.0 | 22 | 1.40 | 0.7 | 0.15 | 1.7×10-17 |
| 41 | 0.20 | 1.0 | 40 | 1.20 | 0.7 | 0.17 | 1.0×10-16 |
| 48 | 0.76 | 0.7 | 53 | 0.59 | 0.7 | 0.18 | 1.0×10-17 |
电荷转移电阻Rct随浸泡时间的变化随浓度不同而不同. 0.01 mol/L
图8所示为低碳钢试样在除氧的4种溶液中长期浸泡至电位攀升到稳定值之后的腐蚀形貌. 结果表明, 在4种环境中样品均产生了明显的腐蚀. 在0.01 mol/L
图9所示为腐蚀产物的XRD分析结果, 发现在所有浓度的
根据文献[29]中Fe-H2O体系的电位E-pH图, 对于重碳酸盐缓冲溶液, 其pH值恒等于8.33. 低碳钢最终的稳定电位处于α-FOOH和Fe3O4的共存区以及α-FOOH稳定区. 所以热力学条件允许α-FOOH和Fe3O4在此种环境下稳定存在.
因为环境中没有O2存在, 所以当腐蚀开始时, 去极化剂为H+, 低碳钢电极的阴极反应为式(1), 其平衡电位为-0.734 V.
根据热力学计算的结果, 下列反应可以自发进行:
结合低碳钢电极在4种不同环境中的开路电位监测曲线(图2), 在浸泡过程刚开始时, 低碳钢电极的开路电位均在-0.81 V左右, 这恰好是图1中极化曲线的自腐蚀电位. 此自腐蚀电位值正是阴极反应(1)式与阳极反应的耦合值, 所以腐蚀初期阳极反应为生成Fe3O4的反应(3). 随着腐蚀产物Fe3O4的不断生成和累积, 导致腐蚀产物在电极表面的覆盖率增大, 电极电位升高. 当电极电位升高超过反应式(4)的平衡电位时, 低碳钢电极表面开始有α-FeOOH生成. α-FeOOH的积累将导致电极电位进一步升高, 当电极电位升高至-0.734 V后, 析氢反应停止. 若假设溶液中Fe2+和Fe3+的浓度为10-5 mol/L, 则α-FeOOH将替代H+作为阴极去极化剂参与腐蚀过程的阴极还原反应[
此时阳极反应仍为反应(3)或(4). 当电极电位升高至高于-0.452 V后, 阳极也可能伴随发生Fe3O4向α-FeOOH转化的如下反应:
图2的开路电位监测曲线显示在0.02, 0.05和0.1 mol/L
根据图2所示的在0.01 mol/L
(1) 低碳钢在无氧
(2) 低碳钢在无氧
(3) 低碳钢在无氧
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|

/
| 〈 |
|
〉 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}