


王新印, 男, 1989年生, 硕士生
应用扫描电化学显微镜的产生/收集和反馈模式, 研究纯Mg在NaCl和Na2SO4溶液中腐蚀过程中的微区(点、线、面)析氢行为和活性点的演变过程. 实验结果显示, 无论在NaCl还是Na2SO4溶液中, 阴极极化和阳极极化均促进析氢, 与传统集气法获得的纯Mg腐蚀过程存在负差数效应一致, 但析氢速率在微区范围内波动加大, 不稳定, 且与极化程度有关. 析氢速率随NaCl浓度增加而增加, 但随Na2SO4浓度增加而减少. NaCl浓度增大、pH值降低、阳极极化有利于Mg表面活性点的产生.
Since electrochemical impedance spectroscopy, polarization curve and hydrogen collection were the main technologies for corrosion research of Mg and its alloy. However, those methods only provide the mean information of entire surface of corrosion electrode. In this work, H2 evolution and active sites of pure Mg from localized sites (point, line and surface) in NaCl and Na2SO4 solution based on generation/collection and feedback modes of scanning electrochemical microscopy (SECM) were studied. The results indicate that both cathodic and anodic polarization are in favor of H2 evolution in NaCl and Na2SO4 solution, which is well in line with the negative difference effect by the classical H2 collection, but the SECM results show that the H2 evolution in localized sites is not uniform and stable. The H2 evolution rate increases with NaCl concentration increasing, which is opposite in Na2SO4solution. The higher NaCl concentration, anodic polarization and lower pH value accelerate the formation of active sites on pure Mg surface.
Mg是一种极其活泼的金属, 其氧化成Mg2+的标准平衡电位为-2.34 V[1], 有很强的失电子倾向. 在自然环境下, Mg表面生成一层类钝化膜, 主要成分为MgO/Mg(OH)2, 但该钝化膜薄而且不致密, 在含有腐蚀性离子(如Cl-)的溶液中或阳极极化[2]下很容易破裂, 裸露出Mg金属基体, 继续发生腐蚀反应. Mg腐蚀的阳极反应为Mg→Mg2++2e-; 阴极反应可分为析氢反应: 2H2O+2e-→H2+2OH-和吸氧反应: O2+4e-+2H2O→4OH-, 由于Mg的标准电极电位远负于析氢反应的标准电极电位, 使得析氢反应以很快的速率发生. 虽然溶液中存在一定浓度的O2, 但O2的还原反应受扩散的控制, 对Mg腐蚀总的阴极行为的影响十分有限. Baril和Pébère[3]利用旋转盘电极研究了Mg在0.1 mol/L Na2SO4中的极化行为, 结果表明, Mg的阴极极化曲线与转速无关, 说明析氢反应是Mg的主要阴极反应, 因为吸氧反应受扩散控制, 不同转速下其扩散速率不同. 因此研究镁合金的腐蚀过程中一般无需考虑O2的还原反应[4].
Mg腐蚀阴极过程的另外一个特点是负差数效应(negative difference effect, NDE)[5], 即在阳极极化下, Mg及其合金析氢速率随着电位的升高而增加, 这有悖于传统电极过程动力学理论. 为了解释Mg的负差数效应, Petty等[6]提出了Mg+机理, Makar和Kruger[7]提出了第二相粒子脱落机理, Song等[2]提出了MgH2机理, Tunold等[8]提出了钝化膜机理, Bender等[9]提出了外电路电子消耗机理, Frankel等[10]提出了析氢交换电流密度增大机理. 但这些机理都存在一定程度的缺陷, 不足以完全解释现有的实验现象. 如Mg+机理和外电路电子消耗机理缺乏直接实验证据; 第二相粒子脱落机理很难解释纯Mg的负差数效应; MgH2机理中生成MgH2是还原反应; 钝化膜机理难以解释镁合金在酸性溶液中的负差数效应; 析氢交换电流密度增大机理从宏观现象出发, 很难解释微观科学机制.
为了定量表征Mg腐蚀过程中的析氢, 经典倒置漏斗排液法[10]是常用的收集方法, 该方法的主要不足有: (1) 只能获得一个时间段的平均产氢量, 而无法得到实时的析氢速率; (2) 产氢量少时, 测量误差大甚至无法测量[11,12]; (3) 收集装置不严密, 容易造成H2的泄露; (4) 体系温度的变化会导致溶液溶解O2和N2量的变化, 溶液在释放或吸收O2和N2的过程中会导致H2体积测量的误差, 例如温度升高, 溶液将释放O2和N2, 造成测得的H2量偏多.
为了从局部微区研究认识镁合金腐蚀过程中的析氢行为, 本工作利用扫描电化学显微镜(scanning electrochemical microscopy, SECM)的产生/收集模式定量表征Mg腐蚀过程中的析氢, 利用反馈模式观察Mg腐蚀过程中微区活性点的产生与变化, 明晰极化电位和阴离子等对Mg腐蚀过程的影响.
高纯Mg中Mg含量大于99.98% (质量分数), 主要杂质为Al, Si和Mn元素. 实验用纯Mg电极尺寸为10 mm×10 mm×2 mm, 实验前分别用400号、600号和800号的SiC砂纸打磨光滑, 再用1 μm的Al2O3抛光膏抛光至镜面, 最后用丙酮除脂, 晾干备用. SECM的探针为Pt超微电极, 直径10 μm, 探针玻璃半径与Pt丝半径比值约为10. 探针在使用前用0.3 μm Al2O3抛光膏抛光至镜面, 并用去离子水清洗, 超声去除表面吸附物. SECM测试采用CHI 920C扫描电化学显微镜系统, 结合双恒电位仪, 软件版本: CHI Version 13.11. SECM测试时采用四电极系统, Pt探针和纯Mg电极为2个工作电极, 2 cm长Pt丝为对电极, Ag/AgCl (KCl浓度为3 mol/L, 相对于标准氢电位为0.194 V)为参比电极. 以下如无特别说明, 所指电位皆相对于此参比电极电位.
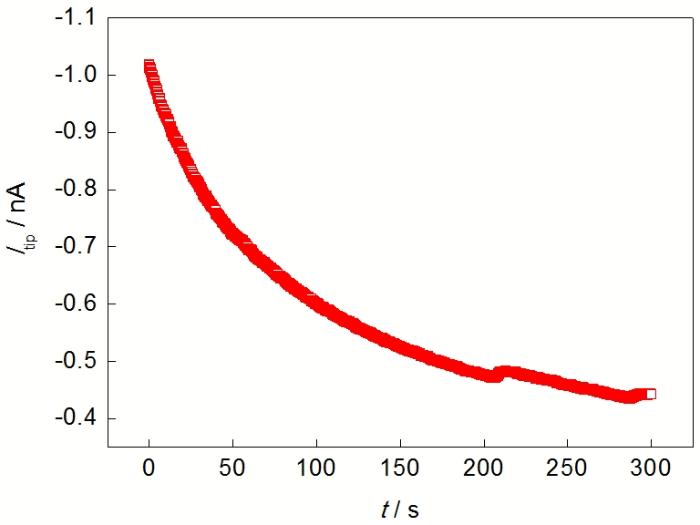
纯Mg置于SECM电解池底端, 用有机玻璃、O型圈和螺丝将其固定, Mg与溶液的接触面积为0.28 cm2. 探针与基底纯Mg之间的距离通过逼近曲线确定, 测试溶液为0.001 mol/L的C11H12FeO (Ferrocenemethanol, FcMeOH)+0.1 mol/L KNO3. 当探针电流等于极限扩散电流70%时, 探针与基底间距离为10 μm. 逼近结束时, 将溶液换成待研究溶液(NaCl或Na2SO4溶液). 浸泡10 min后, 探针电位设为0 V, 收集氧化H2的电流. 因该电位下H2的氧化受扩散控制, 探针的电流仅与H2浓度有关, 而溶液中H2浓度的大小反映了纯Mg的析氢速率. 纯Mg的析氢速率越快, 溶液中H2的浓度越大, 探针氧化H2的电流也越大. 典型的H2收集实验结果如图1所示(图1中纵坐标Itip表示探针电流, 对应H2氧化电流). 可见, 在数据采集的前200 s内, 探针电流下降较快, 主要原因是腐蚀产生的H2与探针消耗H2速率不同, 以及与H2在空间的分布有关; 但是200 s后, 探针电流逐渐稳定, 表明腐蚀产氢和探针氧化H2达到动态平衡. 本工作选取240~300 s的数据, 取其平均值作为探针电流值. 同一条件下的实验重复3次, 对所得结果取平均值及误差. 线扫描实验中探针电位0 V, 扫描距离2000 μm, 面扫描实验的扫描范围200 μm×200 μm, 扫描速度20 μm/s.
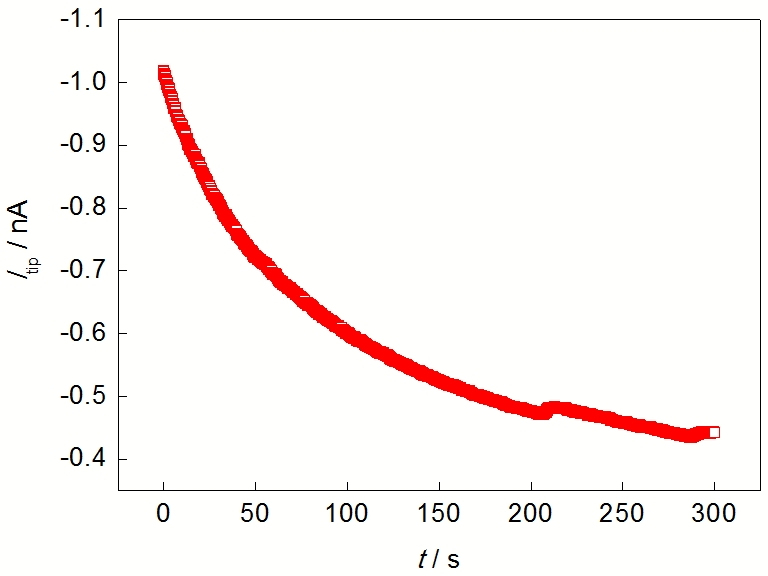 | Fig.1 Typical curve of tip current (Itip) for hydrogen oxidation with time (t)图1 典型探针氧化H2电流与时间的变化曲线 |
{kind=link}
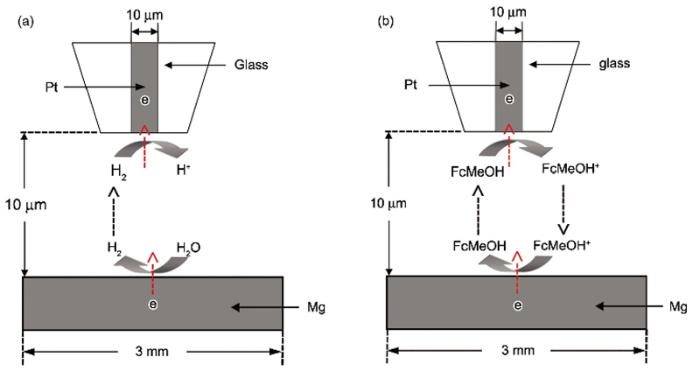
利用SECM反馈模式研究纯Mg表面活性点变化, 探针逼近基底时的溶液为0.001 mol/L FcMeOH+0.1 mol/L NaCl (在探讨Cl-浓度影响时, 为0.5 mol/L NaCl). 逼近结束后, 无需更换溶液, 选取浸泡时间为3, 12和24 h进行面扫描实验, 扫描范围400 μm×400 μm, 扫描速度40 μm/s. 面扫描时探针电位为0.5 V, 在此电位下, FcMeOH的氧化受扩散控制, 探针经过基底活性位置时, 裸露的Mg会将FcMeOH+还原成FcMeOH[13], 对探针电流形成正反馈, 探针的电流反映了Mg表面氧化膜的致密程度及活性点的产生、演变和析氢行为. 产生/收集模式和反馈模式的示意图如图2所示.
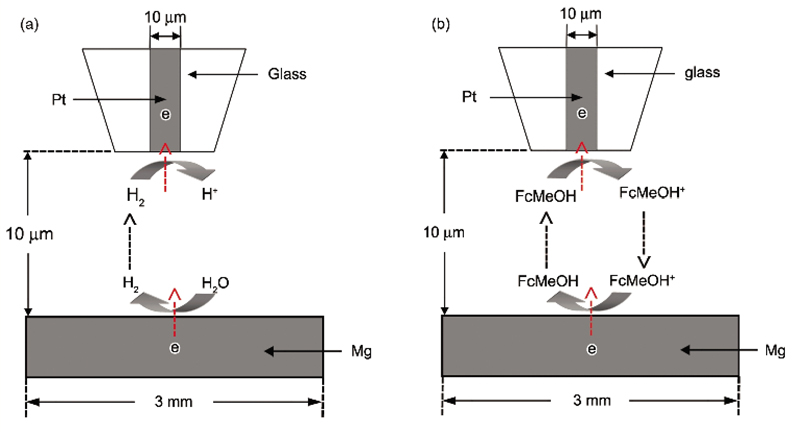 | Fig.2 Schematics of substrate generation/tip collection mode (a) and feedback mode (b) of scanning electrochemical microscopy (SECM)图2 扫描电化学显微镜(SECM)产生/收集模式和反馈模式示意图 |
{kind=link}
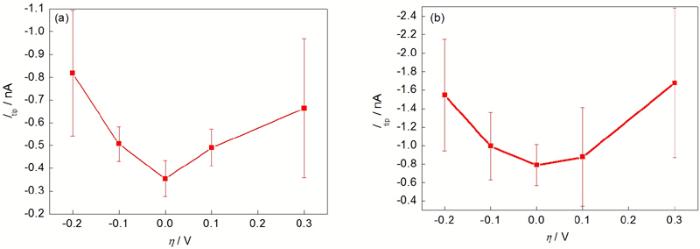
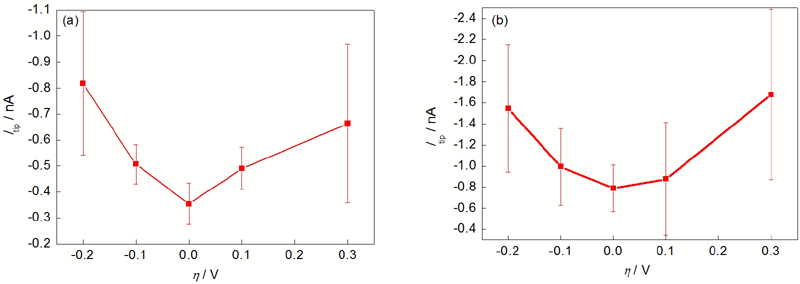 | Fig.3 Tip current for hydrogen oxidation (Itip) of pure Mg under different over potentials ( h) in 0.1 mol/L NaCl (a) and 0.1 mol/L Na2SO4 (b) solution图3 纯Mg在0.1 mol/L NaCl和0.1 mol/L Na2SO4中不同过电位下探针氧化H2电流 |
{kind=link}
分别在0.1 mol/L NaCl和0.1 mol/L Na2SO4溶液中, 进行固定点的H2收集实验. 纯Mg分别处在开路及相对开路-0.2, -0.1, 0.1和0.3 V极化下, 考察极化电位对Mg析氢的影响, 结果如图3所示. 实验结果显示, 无论是在NaCl还是在Na2SO4溶液中, 纯Mg在开路下的析氢速率最小, 阳极极化和阴极极化都促进纯Mg析氢, 表明纯Mg在含Cl-和不含Cl-溶液中均存在负差数效应. 纯Mg在Na2SO4溶液中的析氢电流大于在NaCl溶液中的析氢电流, 表明较短时间内低浓度Cl-对纯Mg腐蚀析氢不具有更强的催化效应. 图3的结果初步证明了利用SECM研究纯Mg析氢的可能性, 但是实验结果也表明, 极化程度较大时, 数据的波动较大, 表明剧烈的析氢反应会导致空间H2分布变化较大, 从而导致探针收集电流波动较大. 另外也表明纯Mg表面析氢不均匀, 这种不均匀性与电极表面有关, 也与极化程度有关.
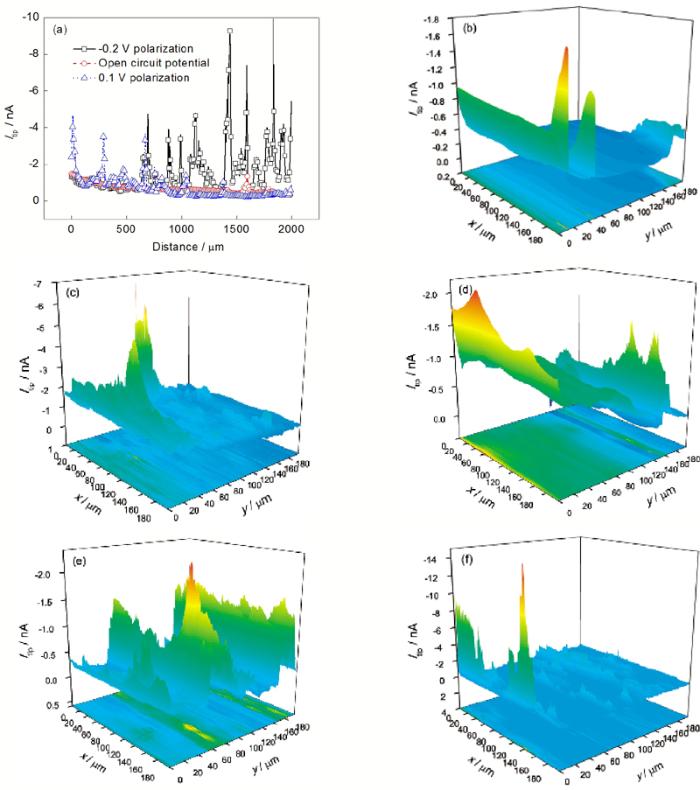
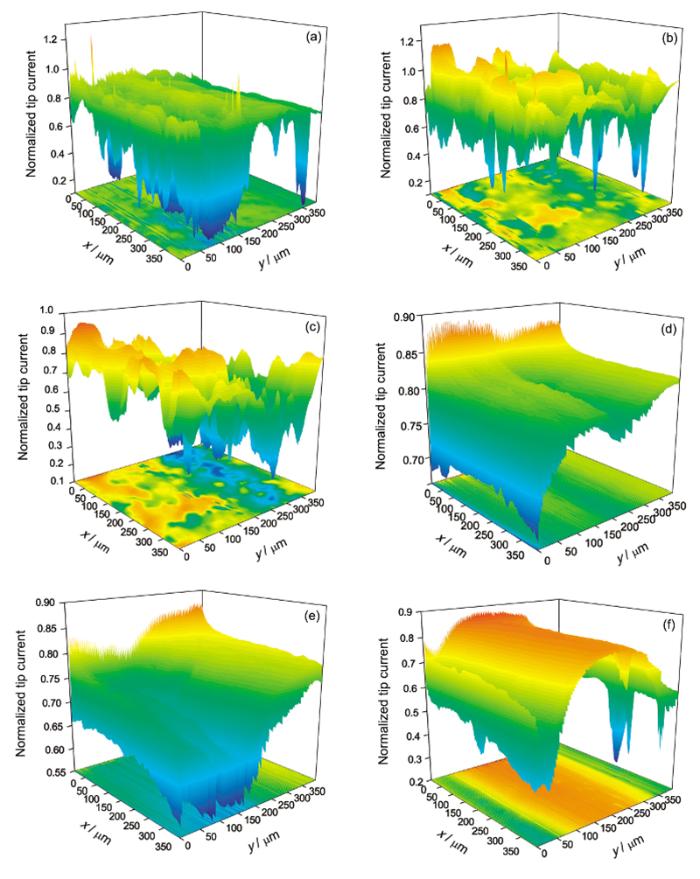
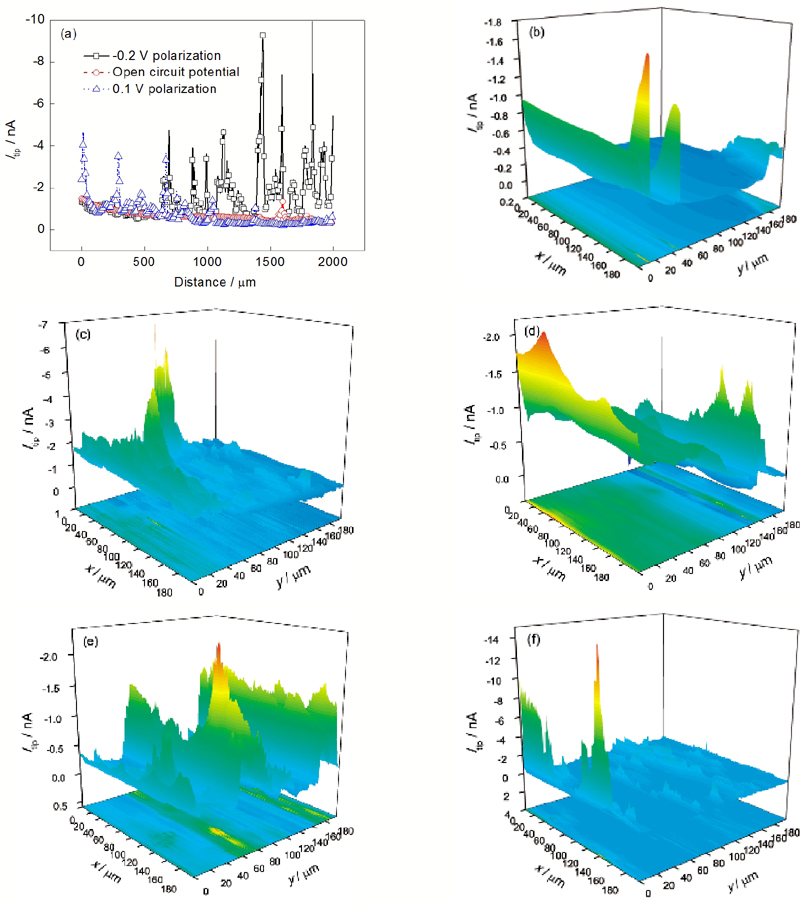 | Fig.4 Line scan results (a) and surface mapping results (b~f) of pure Mg with different polarization potentials in 0.1 mol/L Na2SO4solution(a) line scan results at open circuit potential, -0.2 V and 0.1 V polarization(b) open circuit (c) -0.2 V (d) -0.1 V (e) 0.1 V (f) 0.3 V图4 纯Mg在0.1 mol/L Na2SO4溶液中不同极化电位下的线扫描结果和面扫描结果 |
{kind=link}
在0.1 mol/L Na2SO4溶液中, 纯Mg在开路及相对于开路-0.2和0.1 V极化下的线扫描结果如图4a所示. 在0.1 mol/L Na2SO4中, Mg处在开路及相对于开路-0.2, -0.1, 0.1和0.3 V极化下的面扫描结果如图4b~f所示. 由图可见, 无论是开路、阴极极化还是阳极极化, 纯Mg表面H2的分布都是不均匀的, 且不均匀程度随极化程度的增加而增大. 与图3定点收集氢气的实验结果一致. 图4b~f显示, 在阳极和阴极极化下, 纯Mg表面析氢速率大于开路下, 且存在若干大小不等的电流峰(图4f的电流峰不明显是由于其最大电流峰达到-13 nA, 而图4b~e的最大电流峰均远小于-13 nA). NaCl溶液中的线扫描和面扫描结果与Na2SO4溶液中的结果相似, 即极化使纯Mg表面的析氢增加、空间分布不均匀性加剧.
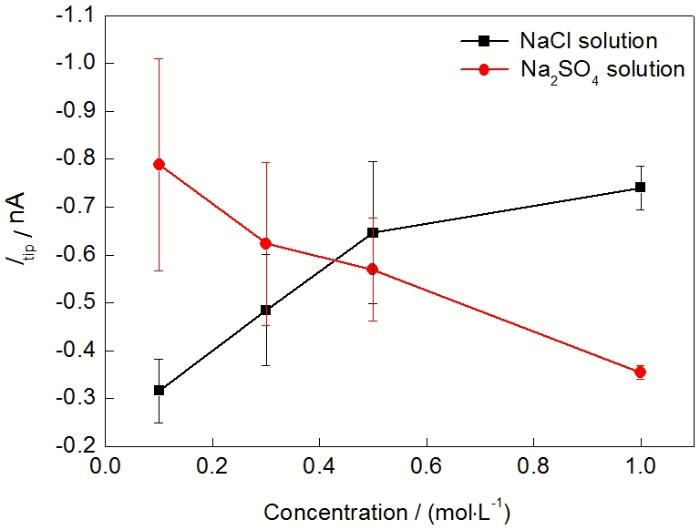
除了外加极化电位, 阴离子种类及浓度对Mg的析氢也有影响. 图5给出了在0.1, 0.3, 0.5和1.0 mol/L的NaCl和Na2SO4溶液中纯Mg的析氢速率. 由图5可知, 纯Mg的析氢速率随NaCl浓度的增大而加快, 随Na2SO4浓度的增大而减慢. 在0.1和0.3 mol/L的低浓度下, 纯Mg在Na2SO4溶液中的析氢速率大于在NaCl溶液中的析氢速率; 而在0.5和1.0 mol/L的高浓度下, 纯Mg在Na2SO4溶液中的析氢速率小于在NaCl溶液中的析氢速率.
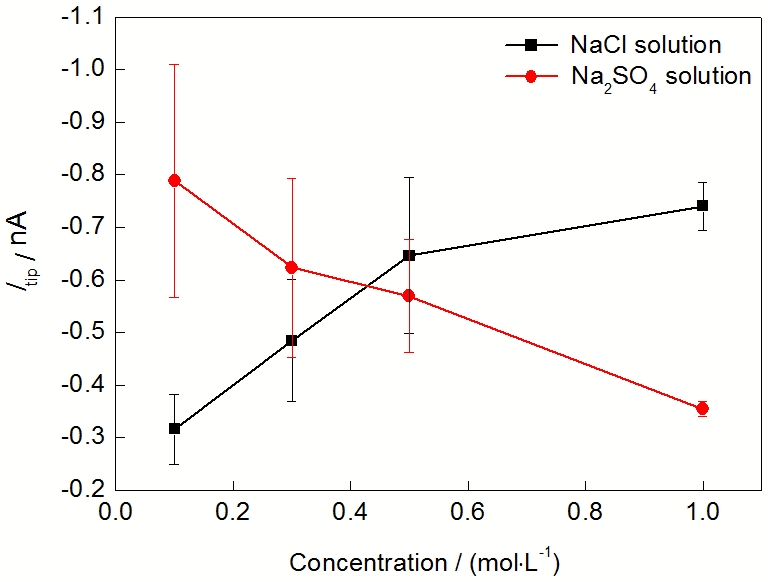 | Fig.5 Tip current for hydrogen oxidation (Itip) of pure Mg in NaCl and Na2SO4 solutions with different concentrations图5 纯Mg在不同浓度NaCl和Na2SO4溶液中的探针氧化H2电流 |
{kind=link}
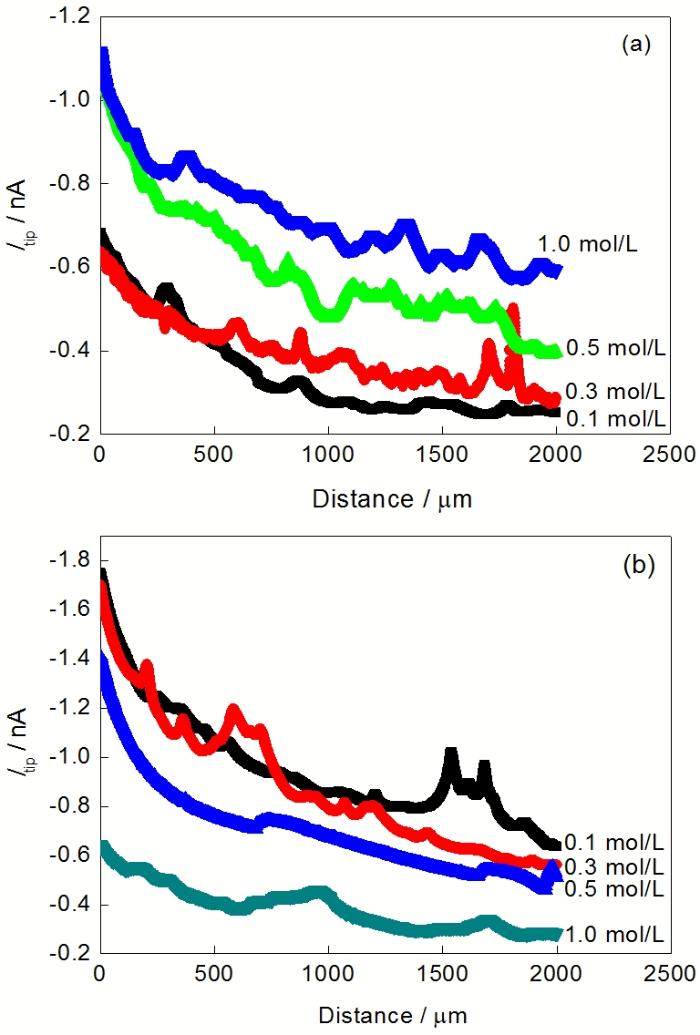
在不同浓度NaCl和Na2SO4溶液中, 纯Mg在开路下的线扫描结果如图6所示. 在NaCl溶液中, Mg的析氢速率随NaCl浓度的增大而加快, 而在Na2SO4溶液中, Mg的析氢速率随Na2SO4浓度的增大而减慢. 不同浓度NaCl和Na2SO4溶液中的线扫描结果再次验证了图4中关于纯Mg表面析氢分布不均匀的结论.
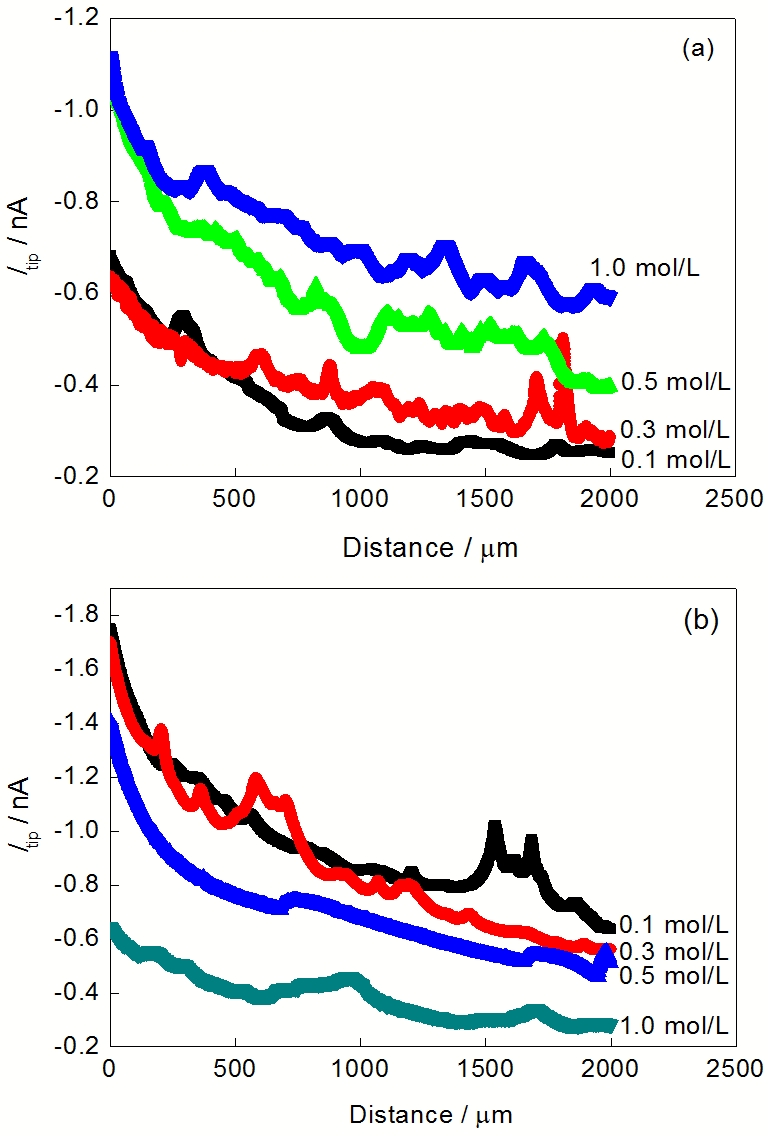 | Fig.6 Line scan results of pure Mg at open circuit potential in NaCl (a) and Na2SO4 (b) solutions with different concentrations图6 纯Mg在开路电位下, 不同浓度NaCl和Na2SO4溶液中线扫描结果 |
{kind=link}
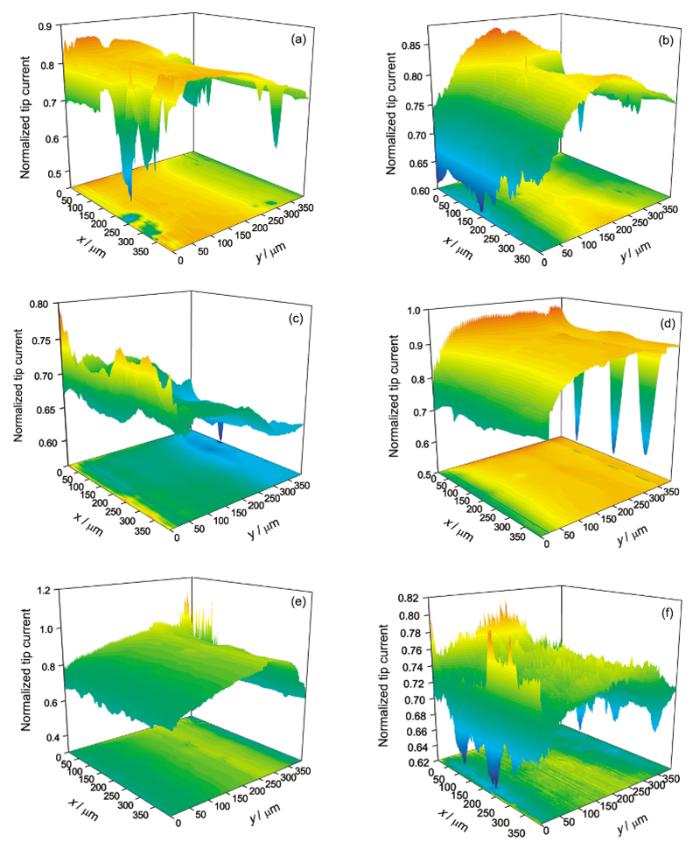
纯Mg在0.1和0.5 mol/L NaCl溶液中浸泡24 h过程中表面的活性点随浸泡时间的变化如图7所示. 图7中的z轴为探针归一化电流, 即探针在面扫描中的电流除以探针离基底无穷远时的稳态电流. 图7a~c的结果显示, 在0.1 mol/L NaCl溶液中浸泡24 h时间范围内, 探针并未出现归一化电流大于1的情况, 即扫描区域并无对探针电流形成正反馈的活性点. 但存在探针归一化电流大于0.7的区域(0.7是探针在10 μm距离时对应的电流比值), 这些区域说明Mg表面的钝化膜在NaCl溶液浸泡过程中已经出现了局部破裂, 而且这些区域随浸泡时间的变化其位置并不固定. 除了探针归一化电流大于0.7的区域, 从扫描结果还可以看到一些探针归一化电流明显小于0.7的区域. 与0.1 mol/L NaCl溶液不同, 在0.5 mol/L NaCl溶液中浸泡12 h时出现了探针归一化电流大于1的区域(图7e), 说明高浓度的Cl-有利于活性点的产生. 但继续浸泡至24 h, 这些活性点消失(图7f), 说明这些活性点可能是亚稳态点蚀, 在Mg继续浸泡过程中, 这些亚稳态点蚀区域又重新钝化.
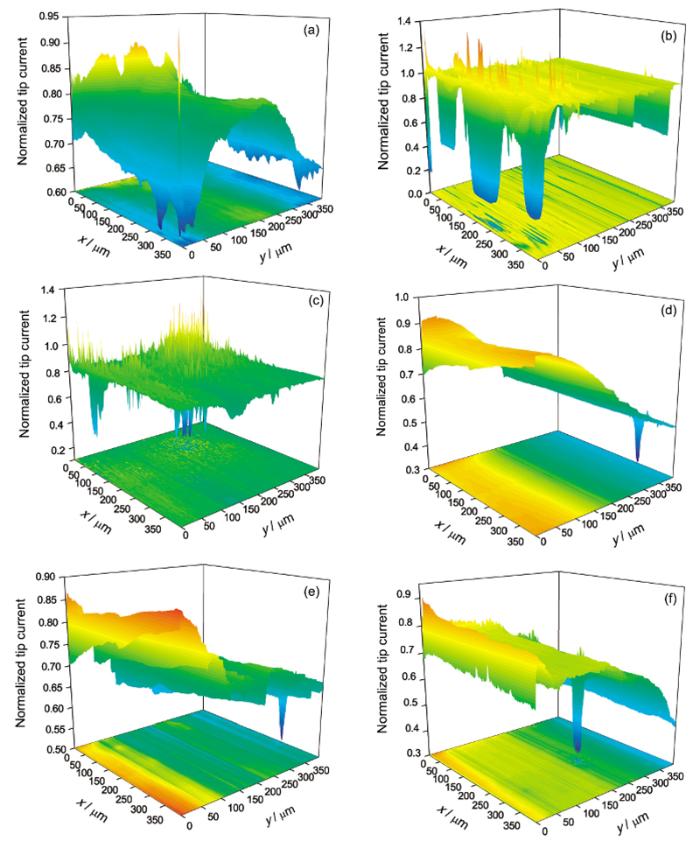
为了研究极化程度对纯Mg活性点产生的影响, 进行了相对于开路电位0.1和-0.2 V极化下纯Mg在0.1 mol/L NaCl溶液中的面扫描测试, 结果见图8. 纯Mg在0.1 V极化12和24 h时, 扫描区域产生了众多的探针归一化电流大于1的区域. 与图7e和f类似, 在浸泡12 h时产生的活性点在浸泡24 h后会消失, 而浸泡12 h时的钝化区域, 至浸泡24 h时可能再次产生活性点. 与阳极极化不同, 当Mg在-0.2 V阴极极化时, 在24 h浸泡时间范围内没有出现探针归一化电流大于1的区域. 比较可以看出, 阳极极化对活性点的产生有促进作用.
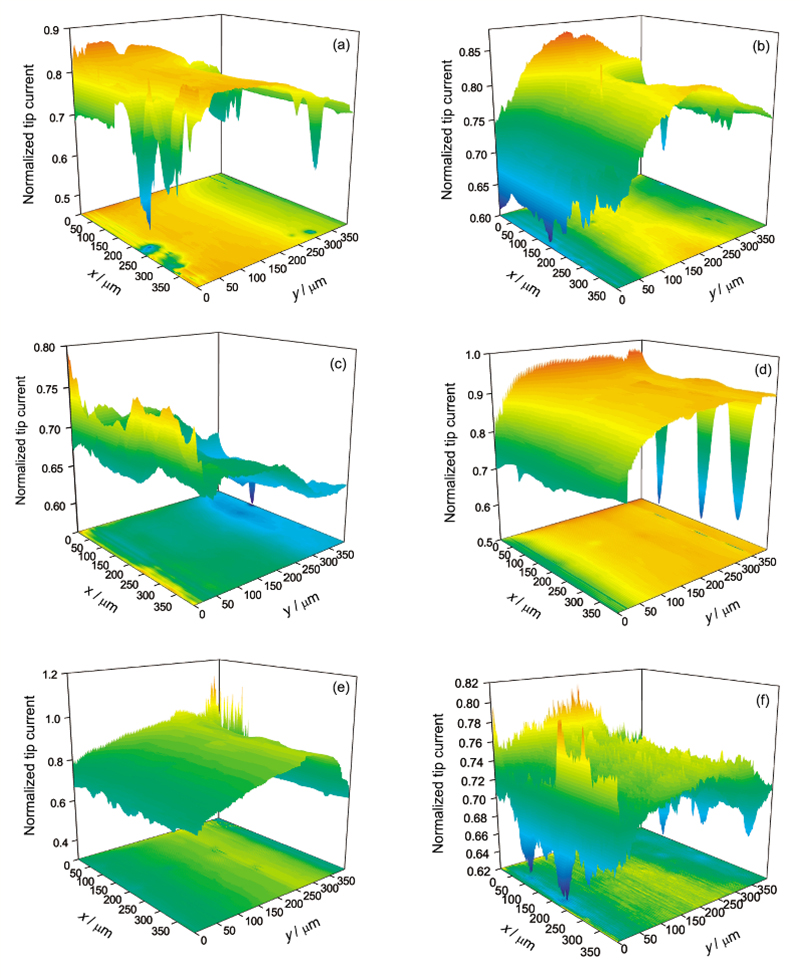 | Fig.7 Surface mappings of pure Mg in 0.1 mol/L (a~c) and 0.5 mol/L (d~f) NaCl solution with 3 h (a, d), 12 h (b, e) and 24 h (c, f)图7 纯Mg在0.1和0.5 mol/L NaCl溶液中不同时间的面扫描图像 |
{kind=link}
溶液的pH值对活性点的产生也会有影响, 在pH=2和11的NaCl溶液中的面扫描结果如图9所示. 在pH=2的NaCl溶液中浸泡3和12 h时, 存在探针归一化电流大于1的活性区域(图9a和b). 虽然图9a和b都有活性区域, 但它们的位置并没有一一对应, 而且图9c中没有观察到探针归一化电流大于1的区域, 再次说明纯Mg表面的活性点是处于不断生成与再钝化的过程. 在pH=11的溶液中, 在整个24 h浸泡时间范围内, 没有发现探针归一化电流大于1的区域. 比较可以看出, 酸性溶液有利于纯Mg表面活性点的产生, 而碱性溶液有利于Mg表面形成钝化膜.
图3和4的结果表明, 纯Mg的极化程度越大, 纯Mg析氢越多, 且H2在纯Mg表面的分布不均匀性增加. 在开路电位下, 析氢反应只能以较小的速率在纯Mg表面发生. 阴极极化减小了析氢反应的活化能, 使析氢反应可以在Mg表面更多的区域以更大的速率发生, 而阳极极化会促使纯Mg表面的钝化膜破裂. 在实验中可以观察到, 随阳极极化时间的增加, 纯Mg表面产生越来越多的黑色区域. 当钝化膜没有破裂时, 析氢反应在钝化膜上发生; 当钝化膜破裂时, 析氢反应主要在这些黑色区域的中心发生[14]. 由于黑色区域主要是MgO, Mg(OH)2和杂质金属的夹杂物, 且粗糙多孔表面积增加, 有利于析氢[15,16,17], 因此析氢反应在黑色区域的反应速率远大于在钝化膜上的反应速率. 阳极极化程度越大, 钝化膜破裂越严重, 产生的H2越多[2].
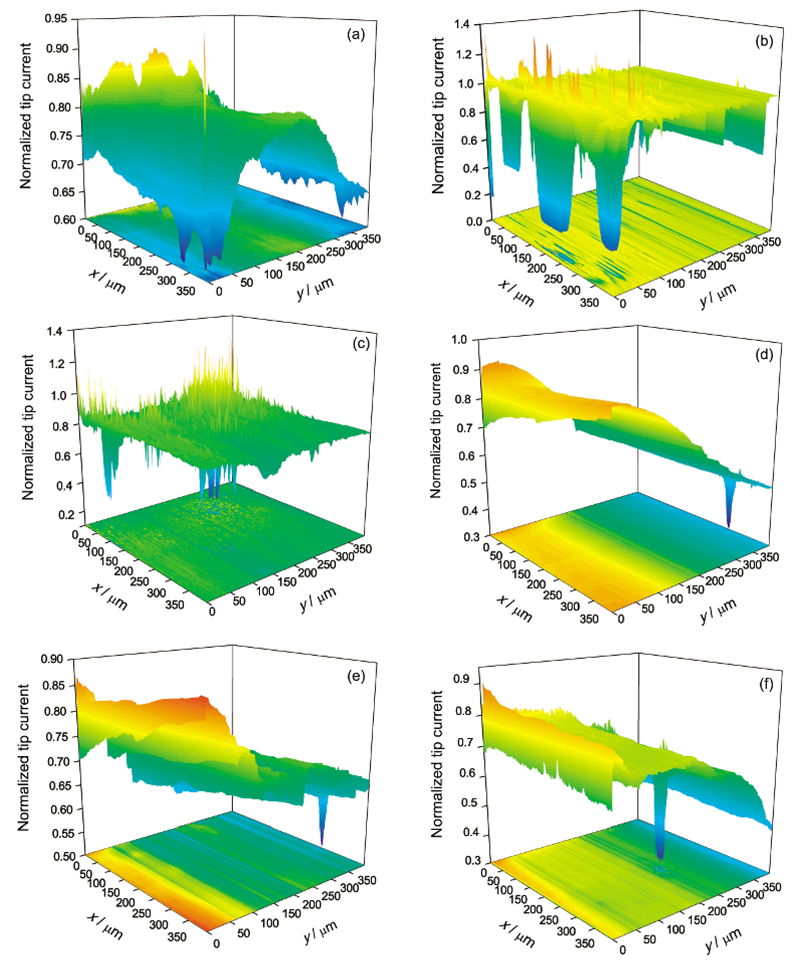 | Fig.8 Surface mappings of pure Mg in 0.1 mol/L NaCl solution at 0.1 V (a~c) and -0.2 V (d~f) polarization with 3 h (a, d), 12 h (b, e) and 24 h (c, f)图8 纯Mg在0.1 mol/L NaCl溶液中不同极化电位、不同时间的面扫描图像 |
{kind=link}
图5和6结果均表明, 随着Cl-浓度的增大, Mg的析氢增多, 与经典集气法研究结果[18]相似. Cl-由于半径较小, 可以较容易地穿透Mg表面的钝化膜[19], 减小钝化膜的致密性和绝缘性, 且Cl-还会与钝化膜的主要成分MgO及Mg(OH)2反应生成MgCl2[15], 破坏钝化膜. Cl-浓度越大, 这种破坏作用越明显, 产生更多的析氢活性点, Mg的析氢越多.
随着
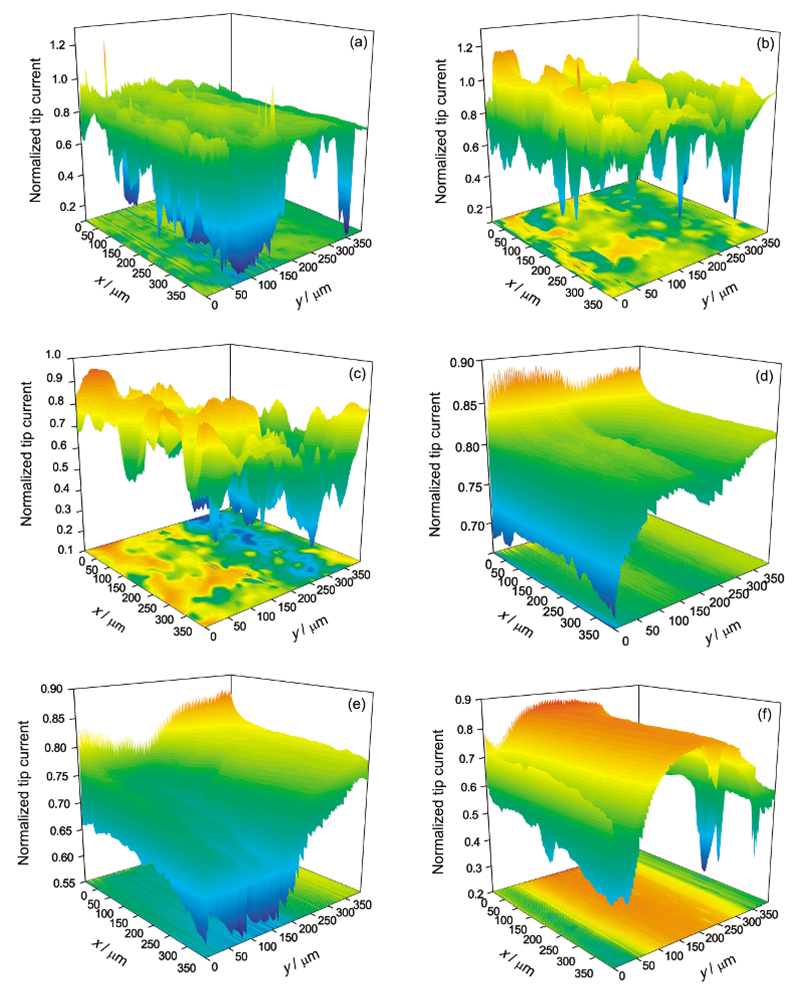 | Fig.9 Surface mappings of pure Mg in 0.1 mol/L NaCl solution at pH=2 (a~c) and pH=11 (d~f) with 3 h (a, d), 12 h (b, e) and 24 h (c, f)图9 纯Mg在0.1 mol/L NaCl溶液中不同pH值、不同时间的面扫描图像 |
{kind=link}
当浓度为0.5和1.0 mol/L时, 纯Mg在NaCl溶液中的析氢速率快于在Na2SO4溶液中, 这是因为Cl-的半径比
图7d~f结果显示, 虽然纯Mg在0.5 mol/L NaCl中浸泡3 h时产生了活性点, 但随后的实验发现此活性点消失, 这可能是由于此活性点为亚稳态点蚀. 由于腐蚀发生过程中的局部阴阳极距离很近, 出现活性点的区域pH值相对较高, 容易重新发生钝化[24]. 反馈实验中, 可以观察到探针归一化电流明显小于0.7的区域, 如图7d中(100, 350), (250, 350), (350, 350) (前一个数代表x轴坐标, 后一个数代表y轴坐标) 3个点. 出现这些区域的原因是H2会以H形式在Mg表面吸附[25,26,27,28,29,30], 进而进入到金属内部, 造成氢脆. 由于H在Mg表面的吸附, 阻碍了FcMeOH向探针的扩散, 所以探针上的电流较小.
比较图7a~c和d~f可以看到, 在0.1 mol/L NaCl中, 整个扫描时间范围内, 没有发现活性点, 而在0.5 mol/L NaCl中浸泡12 h, 观察到了活性点, 虽然此活性点在随后的浸泡过程中消失, 但在0.5 mol/L NaCl中, 探针的整体电流要大于0.1 mol/L NaCl中, 说明提高Cl-浓度, 对纯Mg表面钝化膜的破坏加剧, 有利于活性点的产生. Cl-浓度对Mg合金的腐蚀阳极溶解行为影响较大.
比较图7和8, 极化电位在0.1 V时, 浸泡时间为12和24 h时, 纯Mg表面产生了明显的活性点, 而在-0.2 V极化下, 在浸泡24 h时间范围内, 没有观察到活性点的产生, 说明阳极极化有利于纯Mg表面活性点的产生. 纯Mg在阳极极化时, 表面会有更多的活性点用于点蚀成核. 阳极极化也会增加钝化膜的电场强度, 加强对Cl-等腐蚀性阴离子的吸附[21], 导致钝化膜更容易破裂, 产生活性点, 而阴极极化时产生的OH-则有利于钝化膜的生成和稳定.
对比图7和9, 可以发现在pH=2的溶液中, 浸泡时间为3和12 h时, 纯Mg表面出现了活性点, 而在pH=11的溶液中, 在整个浸泡时间范围内, 没有发现活性点的产生. Tian等[19]发现AZ91D镁合金在不同pH值的Na2SO4溶液中, 腐蚀电位随pH值减小而变负. Song和Atrens[31]认为pH>10.5时, Mg不易发生腐蚀. 这些实验结果都说明低pH值有利于Mg表面活性点的产生而高pH值有利于钝化膜的生成.
(1) 利用扫描电化学显微镜的产生/收集模式定量研究了纯Mg在不同极化、不同浓度NaCl及Na2SO4溶液中的析氢行为及其空间分布. 实验结果表明, 纯Mg的极化程度越大, 其析氢速率越快, 且H2在纯Mg表面的分布越不均匀. 纯Mg的析氢速率随NaCl浓度的增大而加快, 随Na2SO4浓度的增大而减慢. 纯Mg在低浓度Na2SO4溶液中的析氢速率快于其在相同浓度NaCl溶液中的析氢速率, 而在高浓度溶液中则相反.
(2) 利用扫描电化学显微镜的反馈模式研究了纯Mg在不同浓度NaCl溶液、不同极化程度及不同pH值下活性点的产生与演化情况. 镁合金的腐蚀行为以非稳态局部腐蚀为主, 腐蚀活性点生成、再修复过程交替出现. 增大NaCl浓度、阳极极化及降低溶液pH值都对纯Mg的钝化膜有破坏作用, 促进纯Mg表面活性点的产生.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|