
陈浩, 张璁雨 , 朱加宁, 杨泽南, 丁然, 张弛, 杨志刚
, 朱加宁, 杨泽南, 丁然, 张弛, 杨志刚
清华大学材料学院教育部先进材料重点实验室 北京 100084
CHEN Hao, ZHANG Congyu, ZHU Jianing, YANG Zenan, DING Ran, ZHANG Chi, YANG Zhigang
文献标识码: TG11, TG142
文章编号: 0412-1961(2018)02-0217-11
通讯作者:
收稿日期: 2017-11-6
网络出版日期: 2018-02-20
版权声明: 2018 《金属学报》编辑部 《金属学报》编辑部
基金资助:
作者简介:
作者简介 陈 浩,男,1986年生,助理教授,博士
展开
摘要
相变是钢铁材料微观组织调控的关键手段。高性能化的发展需要对钢铁材料组织进行更加精细的调控,这也对相变理论的认知提出了更高的要求。奥氏体-铁素体相变是先进高强钢制备过程中最为重要的相变之一。相变过程中的界面迁移与元素配分行为在很大程度上决定了钢铁材料制备过程中组织的演化过程,对实现微观组织的精细化调控至关重要,一直以来是钢铁相变领域的研究热点与难点。本文从理论模型和实验研究2方面,简要综述了近年来国内外关于奥氏体-铁素体相变的界面迁移与元素配分行为的研究进展,并对该研究方向尚未解决的科学问题进行了讨论与展望。
关键词:
Abstract
Phase transformation is one of the most effective methods to tailor microstructure of steels. In order to develop high performance steels, microstructure has to be precisely tuned, which requires a deep understanding of phase transformation. The austenite to ferrite transformation in steels has been of great interest for several decades due to its considerable importance in the processing of modern high performance steels, and it has been investigated from various aspects. Mechanism of interface migration and alloying elements partitioning during the austenite to ferrite transformation was regarded as one of the most significant and challenging topics in the field. This paper briefly summarized the recent progress in the understanding of this topic from both theoretical and experimental perspectives, and would also provide discussions and outlook of the unresolved issues.
Keywords:
固态相变是金属材料调控微观组织和改善性能的关键手段之一。与其它金属材料相比,钢铁材料具有丰富的相变行为,其微观组织和性能具有更高的可调控性,因此应用极为广泛。近年来,钢铁材料高性能化的实现主要依靠对微观组织进行更加精细的设计,这也对组织调控理论的认知提出了更高的要求。奥氏体(γ)-铁素体(α)相变是先进高性能钢制备过程中最为重要的相变之一,也是目前为止研究最多的一种相变。奥氏体-铁素体相变包括形核与长大2个过程。形核过程极为复杂且难以准确观测,经典形核理论很难被实验验证,因此研究进展缓慢[1,2,3,4];相对而言,对Fe-C、Fe-M、Fe-C-M等合金系 (M=Mn、Ni、Si、Cr等置换型元素)中的铁素体长大过程则进行了大量的理论和实验研究[5,6,7]。铁素体长大伴随着2个重要的物理过程:α/γ界面迁移和元素配分。界面迁移,即fcc结构的奥氏体和bcc结构的铁素体之间的转变,是一个能量消耗的过程,能量消耗的大小取决于界面迁移率。从热力学角度上讲,在界面迁移的同时,元素应在两相间重新配分,从而降低体系的自由能。然而从动力学角度上讲,元素是否会配分不仅取决于热力学同时还会受界面迁移速率的影响。与界面迁移一样,合金元素配分行为也会导致能量消耗,进而影响相变动力学。奥氏体-铁素体相变过程中的界面迁移和元素配分的耦合机制一直以来是固态相变领域的研究热点和难点, 对它的深入研究可以为钢铁材料的成分和工艺设计提供重要的理论基础。
近年来,受益于实验表征技术(场发射电子探针分析(FE-EPMA)、激光扫描共聚焦显微镜(LSCM)、扫描透射电子显微镜能谱(STEM-EDX)、电子能量损失谱(EELS)、同步辐射高能X射线衍射(synchrotron X-ray tomography)及三维原子探针(3DAP)等)和多尺度计算模拟手段(第一性原理、元胞自动机、相场等)的发展,学术界对奥氏体-铁素体相变中界面迁移和元素配分过程的认知不断深入,但依然存在诸多尚未解决的争议问题。本文简要综述该领域近些年来的理论模型及实验研究进展,同时对尚未解决的科学问题进行讨论和展望。
根据界面迁移和元素配分的特点,铁素体长大的理论模型可分为3类:(1) 界面控制长大模型[1],即相变过程中不发生元素的配分行为,界面的迁移速率完全取决于界面迁移率和驱动力;(2) 扩散控制长大模型[8],即认为界面迁移率无限大,而不对整体相变动力学产生影响,因此相变由元素在两相间的扩散所控制;(3) 混合控制长大模型[9],即认为整体相变动力学是由界面迁移和元素扩散2个过程共同控制。
界面控制长大的概念由Christian[1]在上世纪70年代提出,认为界面迁移速率与界面迁移率具有如下关系:
式中,v为界面迁移速率;Mint为界面迁移率,是温度的函数;ΔG为新相和母相的自由能差值;R为气体常数;T为热力学温度;M0为常数;Q为原子激活能。
当过冷度或过热度较大,即ΔG
而当过冷度或过热度较小,即ΔG
式(4)常被用于描述二元合金的等温块状相变的动力学过程[11,12]。其中,相变驱动力仅被界面迁移过程消耗,即界面迁移速率与相变驱动力成正比,比例系数与界面迁移率有关。
扩散控制长大模型[8]认为界面迁移过程完全由元素扩散控制,即界面处晶体结构改变而消耗的能量可以忽略,界面迁移率无限大。该模型的核心思想是确定铁素体-奥氏体的界面条件,通过将其与Fick扩散方程及物质守恒方程联立,解得界面迁移的动力学。Zener[8]在上世纪40年代提出的扩散控制生长模型在二元合金体系得到了很好的应用。然而,对Fe-C-M三元体系(M=Mn、Ni、Si、Cr等置换型元素),界面条件的确立则复杂得多。从热力学上讲,C和M会在新相和母相间重新配分以减小体系的自由能;但从动力学上讲,由于M的扩散系数比C的扩散系数小4~5个数量级,因此实际相变过程中合金元素是否会在两相间发生配分仍存在很大争议[6,13~18]。根据合金元素配分情况的不同,不同学者分别提出了准平衡(paraequilibrium,PE)模型[13,14]和局域平衡(local equilibrium,LE)模型[19,20,21,22,23,24,25,26]。
PE模型认为界面处仅C原子处于平衡状态,而合金元素在新相和母相之间不发生配分,界面迁移完全由C扩散控制。其界面条件如下式所示[14,16]:
式中,i=0表示Fe元素;i=1表示C元素;i≥2表示置换型合金元素;
而LE模型认为各元素在界面处均达到局域平衡,即各元素在界面两侧化学势相等[23,24,25,26]:
在过冷度较低时,C和M需在新相和母相间重新配分以减小体系的自由能,然而M扩散系数远小于C,即铁素体生长受合金元素扩散控制,因此相变速率极慢,生长几乎停滞。这种相变模式称为配分局域平衡(partition local equilibrium,PLE)模式;在过冷度较大时,新相可继承母相的M含量,即铁素体长大无需M发生长程配分,铁素体生长受C扩散控制,相变速率较快。这种相变模式称为不配分局域平衡(negligible partition local equilibrium,NPLE)模式。PLE和NPLE模式之间的转变温度称为PNTT (PLE/NPLE transition temperature)。PLE和NPLE模式下界面附近C和M的配分情况如图2所示。类似地,对于同一等温截面,当体成分发生变化时,同样会引起NPLE和PLE之间的模式转换。
以等温奥氏体-铁素体相变为例,在对等温温度进行选择时,可通过合金成分及相关热力学数据计算得到PNTT[27]。在PNTT以上,相变以PLE模式进行,由于铁素体生长受合金元素扩散控制,速率极其缓慢,因此很难得到大量的相变产物;而在PNTT以下,相变则呈现快速的NPLE模式。由于奥氏体基体并非无限大,随着相变的进行,基体中的C富集程度不断增加,当基体中C浓度达到PLE/NPLE转变线时,相变过渡至PLE模式。由于此后相变速率极慢,铁素体体积分数几乎不再增加,转变过程进入一个近乎稳定的状态,即准稳态。故准稳态时的铁素体体积分数可近似认为是该温度下所能得到的最大体积分数。整个过程中铁素体体积分数随时间的变化关系可通过联立界面条件方程(式(6))及物质守恒方程求解得到[28]。
图1 Fe-C-M三元系准平衡(PE)示意图
Fig.1 Fe-C-M ternary alloy under paraequilibrium (PE) condition(a) isothermal section(b) alloying element profile at the α/γ interface(c) C profile at the α/γ interface. Here, XC is the mole fraction of C and YM is the site fraction of alloying element (YM=XM/(1- XC), XM is the mole fraction of alloying element)
图2 Fe-C-M三元系局域平衡(LE)示意图
Fig.2 Fe-C-M ternary alloy under local equilibrium (LE) condition(a) isothermal section in partition local equilib- rium (PLE) mode(b) alloying element profile at the α/γ interface in PLE mode(c) C profile at the α/γ interface in PLE mode(d) isothermal section in negligible partition local equilibrium (NPLE) mode(e) alloying element profile at the α/γ interface in NPLE mode(f) C profile at the α/γ interface in NPLE mode
需要注意的是,尽管PE和NPLE模式下C均在界面处达到局域平衡,M在铁素体和奥氏体内亦不发生长程扩散,界面迁移速率均受C扩散控制。但是,由于NPLE模式下M在界面附近的短程配分将很大程度上改变C在界面处的活度,因此会极大地影响界面迁移的动力学过程。从模拟结果而言,PE模式下的界面迁移速率和准稳态下的铁素体体积分数均高于NPLE模式。
1.3.1 Fe-C二元混合生长模型 与扩散控制长大模型不同,混合控制长大模型[9]假设界面迁移率并非无限大,实际相变动力学应是界面迁移和元素配分共同控制的结果。
Krielaart等[9]在上世纪90年代发现传统的扩散控制生长模型并不能很好地解释Fe-C二元系中γ→α相变过程,并率先提出了混合控制生长的概念,同时考虑C扩散和界面迁移率对铁素体长大动力学的影响,发现C在界面处的浓度分布将同时取决于其扩散速率和界面迁移速率。在Krielaart等的混合生长模型基础上,Sietsma和Van der Zwaag[29]提出了一种简洁的等温混合控制生长模型,认为铁素体的生长模式是介于界面控制生长和扩散控制生长的一种中间状态。他们预测实际相变应是由界面控制生长模式开始,并向扩散控制生长模式演化的过程,界面迁移速率表达式如下:
式中,χ是化学驱动力比例常数;
1.3.2 基于溶质拖曳理论的多元混合生长模型
上述模型中,相界面被认为是纯粹的数学界面,不具有宽度和特定结构,无法容纳溶质原子。实际上,由于溶质原子与Fe原子在尺寸、电负性等元素特性上存在差异,溶质原子会倾向于偏聚到比临近晶粒内混乱度更高的界面处,从而降低体系自由能。溶质原子与移动界面间会产生相互拖曳的作用,即溶质拖曳效应。目前,对溶质拖曳效应的实验研究较为少见,通常以理论模拟来估算其强弱。从拖曳力的角度,文献[31,32,33]最早在上世纪中期提出了溶质拖曳理论,解释了溶质原子对晶界迁移的影响。之后,Purdy等[34]和Enomoto[35]将溶质拖曳模型扩展至Fe-C-M三元系相界面,假设相界面内存在楔形的能量势阱,其中势阱深度与溶质-界面的结合能E0有关,E0的绝对值越大表示溶质元素向界面内偏聚的倾向越强烈。溶质拖曳理论发现当界面在高速和低速移动时,溶质拖曳效应并不显著,最大的溶质拖曳力出现在中速阶段。与溶质拖曳模型不同,Hillert和Sundman[36]从能量角度出发,考虑元素在界面内扩散造成的能量消耗,并从理论上解释了模型与文献[31,32,33]的模型在一定条件下的等效性。之后,也有研究对界面做出不同的假设处理,如离散化[37]或假设为热力学性质连续变化的界面[38]。
Chen等[39,40]基于溶质拖曳理论,考虑合金元素在界面内的偏聚及晶粒内的软碰撞效应,提出了更具普适性的界面处自由能守恒模型(GEB),用于描述Fe-C-M三元系中的γ→α相变动力学。研究表明,二元体系的混合控制生长模型及传统PE、LE模型均可看作GEB模型的特殊情况。对于三元以上合金体系,如Fe-C-Mn-Si,通常认为Mn和Si之间的吸引作用使这2种组元与界面之间具有耦合溶质拖曳效应[41,42,43]。然而也有研究[44]表明,Mn和Si在界面处的溶质拖曳效应并不是简单地叠加结果,而是存在反耦合溶质拖曳效应[44,45]。
溶质拖曳模型能够较好地评估不同置换型合金元素对界面迁移的影响,为铁素体相变速率及相体积分数低于经典模型预测值的现象提供了系统的理论解释,认为这种偏差源自于元素与界面的相互作用。目前,诸多基于溶质拖曳理论的混合生长模型可以用于描述铁素体偏离准稳态的生长过程。然而模拟结果会受到相关界面参数选取的影响,如界面结合能和溶质穿过界面的扩散系数等,如何准确获得这些重要物理参数将是今后研究重点。
1.3.3 相场模型 相场理论[46,47]是基于Ginzburg-Landau自由能泛函理论构建的,可将基体和界面以连续整体的方式处理,用连续的方式考虑体系自由能降低的动力学过程,可用于研究非平衡态微观组织演化。相场理论引入了序参量(或称为相场变量)来描述体系任意时刻任意位置的所属域,通常相场变量在基体区域为固定值,而在弥散界面内连续变化。早期相场法主要用于研究合金中的凝固过程[48],近年来同样被应用于钢中的固态相变[49],目前已成为模拟材料介观尺度组织演变最有力的工具之一。
相场模型[50]同时考虑了界面迁移率和元素扩散(包括软碰撞效应)等因素,是一种混合控制生长的模型。将相场理论用于模拟γ→α相变,需要求解序参量随时间变化的演化方程(即Allen-Cahn方程[51],式(9))与溶质浓度随时间变化的演化方程(即Cahn-Hilliard扩散方程[52],式(10))的非线性方程组,进而描述每一时刻每一位置的状态:
式中,ϕ是某一时刻某一位置的序参量,t为时间,F是系统总能量,L是与界面迁移率有关的参数,c是某一时刻某一位置的某溶质成分,Mc是与原子迁移率有关的参数。由于自由能方程形式以及界面条件选取的不同,研究者提出了不同形式的相场模型。其中,Steinbach等[53,54]提出的多相场模型与界面迁移率和界面能等物理量建立了较为简单明晰的关联而被广泛应用[55,56,57,58]。该模型可描述多晶粒系统相变过程,每个晶粒用单独的序参量表示。对于N个晶粒体系,有N个序参量ϕi (i=1,
式中,Mij是晶粒i和晶粒j之间的界面迁移率,σij是界面能,ηij是界面厚度,ΔGij是驱动力。
目前,钢中奥氏体-铁素体相变的相场模拟通常局限于二元合金体系[60,61,62],这是由于M和C的扩散系数存在4~5个数量级的差距,导致在相同时间尺度下的扩散距离存在巨大差异,对相场模型的计算效率形成了较大的挑战。在近期的一些研究[56,63,64]中,将多元体系简化为伪二元系进行处理,即假设准平衡界面条件,只有C在两相间发生配分,而不考虑M的扩散。由于未考虑M在界面处的偏聚和配分对奥氏体/铁素体界面迁移的影响,基于准平衡假设的相场模型计算结果通常与实验结果偏离。为了解决这一问题,Chen等[65]和Zhu等[66]在相场模型中引入界面处自由能守恒的概念,从而在相场模型中考虑了合金元素在界面内扩散造成的能量消耗,成功模拟了Fe-C-Mn合金循环相变过程中的铁素体长大滞缓现象。张军等[67]则进一步讨论了Mn含量对微观组织形貌和C浓度的影响,强调了M界面短程配分行为在整体相变过程中的重要性。
由于C的扩散较快,且在铁素体中固溶度极低,因此Fe-C合金中等温奥氏体-铁素体相变过程通常伴随着C从铁素体向奥氏体扩散行为。Bradley等[68]对一系列Fe-C合金开展了等温奥氏体-铁素体相变实验,通过金相观察测得了铁素体厚度和长度方向上的长大速率,其实验结果随后被Krielaart等[69]利用二元混合控制模型进行了模拟,发现当相变过冷度较小时,铁素体长大主要受C扩散控制;而当过冷度较大时,相变动力学则决定于界面反应的速率。Liu等[70]则通过膨胀仪测量了Fe-C二元合金在连续冷却和等温过程中的奥氏体-铁素体相变动力学,并运用界面控制和扩散控制生长模型拟合实验测得的相变动力学曲线,推测相变过程中存在由扩散控制到界面控制的模式转换。
扩散控制到界面控制的相变模式转变同样存在于Fe-M二元合金的奥氏体-铁素体相变过程中。但是由于置换型合金元素的扩散极其缓慢,因此在实际过程中,扩散控制型相变很难被观测到。Fe-M合金将在到达某一临界温度后直接发生界面控制型相变,也被称为块状相变,其界面迁移速率处于10-6~10-5 m/s量级且在相变过程中基本保持不变[12,71]。尽管对于块状相变的定义仍存在较大争议,但是块状相变的以下2个特点已得到广泛认可[72]:(1) 新相和母相之间没有特定的取向关系;(2) 新相和母相具有相同的成分。鉴于新相和母相的化学成分相同,Fe-M合金中块状相变发生的热力学极限温度应为T0,此温度下新相和母相具有相同的自由能。然而,Borgenstam和Hillert[73]利用扩散偶实验研究了Fe-Ni合金中块状相变的临界温度,并发现临界温度低于T0。Zhu等[74]运用热膨胀仪测量了Fe-Ni、Fe-Mn和Fe-Co二元合金连续冷却和连续加热过程中奥氏体-铁素体相变以及铁素体-奥氏体相变行为,发现奥氏体-铁素体块状相变临界温度通常低于T0,而铁素体-奥氏体块状相变的临界温度则高于T0。同时实验还发现随着Mn和Ni含量的升高,Fe-Mn和Fe-Ni合金的块状相变临界温度与T0的偏离程度增大,而Fe-Co合金的块状相变临界温度则始终接近T0。因此块状相变临界温度和T0之间的偏差与合金元素种类以及浓度直接相关,如图3[74]所示。这也为溶质拖曳理论的预测[74,75]提供了间接证据:一方面,合金元素在界面处的偏聚和短程扩散产生溶质拖曳效应,使得相变的发生需要更大的驱动力,导致块状相变的相变点与T0偏离;另一方面,溶质拖曳效应的强弱会受到不同种类溶质原子的溶质-界面结合能(E0)的影响,图3[74]呈现出
除了相变模式转变机制,研究Fe-M合金中奥氏体-铁素体相变的另外一个重要目的是获得奥氏体/铁素体界面迁移率。界面迁移率是材料相变的关键物理量,也是所有相变动力学模型必需的物理参数。因缺乏相关数据,在较长的一段时间内Speich等[77]测定的铁素体晶界迁移率被用来近似奥氏体/铁素体界面迁移率。之后Krielaart等[78]和Wits等[79]测量了一系列Fe-M二元合金奥氏体-铁素体相变动力学,通过模型与实验的拟合获得奥氏体/铁素体界面迁移率,结果均小于上述铁素体晶界迁移率。Hillert等[80]对Liu等[81,82]利用膨胀仪测得的实验数据进行研究,认为奥氏体/铁素体界面迁移率比铁素体晶界迁移率小若干数量级。
奥氏体/铁素体界面迁移率的测量结果通常存在量级上的差异,这对定量模拟奥氏体-铁素体相变动力学带来了很大的挑战。最近Zhu等[74]利用高精度膨胀仪对Fe-M合金中奥氏体-铁素体相变及其逆相变动力学进行了系统的实验研究,结果表明,奥氏体/铁素体界面迁移率与合金成分、合金元素种类及相变方向无关,是一种界面本征物理量,如图4[74]所示。
由于同时存在C和M 2类元素,且它们的扩散系数存在巨大差异,铁素体的生长可能会呈现如前所述的多种长大机制。
PE模型和LE模型最常被用来与实验结果进行比较。诸多学者从奥氏体-铁素体准稳态阶段铁素体体积分数、未转变奥氏体中C含量[83,84]及相变动力学发生转变时的临界温度[27,85]等方面进行了研究。结果表明,在大部分情况下LE模型的预测结果优于PE模型。与电子探针分析(EPMA)相比,FE-EPMA可以在更精细的尺度实现对局部C含量较为准确的测定,其束斑大小在100 nm左右,且C含量误差可控制在0.03% (质量分数)左右[86]。Liu等[83]借助FE-EPMA测定了Fe-xC-2Mn (质量分数,%)三元合金在相变进行至准稳态阶段时奥氏体中的C含量,同时用金相法统计了准稳态阶段的铁素体体积分数,发现二者均与LE模型预测相符,且显著偏离平衡相图及PE模型预测结果。刘志远等[85]对Fe-0.073C-2.17Mn-0.80Si-0.88Cr (质量分数,%)合金进行金相组织观察,根据不同温度下等温相同时间所得的铁素体体积分数,发现存在明显的相变动力学转变温度,且与LE模型的预测结果吻合较好。其后,Zhang等[27]同样发现,在一系列Fe-C-Mn、Fe-C-Ni及Fe-C-Mn-Si等低合金钢中,LE模型可以更好地预测其缓慢冷却铁素体相变温度Ar3。
以上实验结果表明,LE模型的预测结果在多数情况下优于PE模型,能够定性地描述Fe-C-Mn、Fe- C-Ni、Fe-C-Mn-Si等合金体系的奥氏体-铁素体相变动力学,因此可以利用1.2节中的部分理论对工业生产进行初步的指导。尽管如此,LE模型仍然存在诸多尚未解决的问题:(1) 研究[83,87,88]发现,在等温铁素体相变中会存在PE至LE的模式转变,实际铁素体相变动力学并不能单一地用LE或者PE理论解释;(2) LE模型中,在界面两侧合金元素达到局域平衡时,在奥氏体母相一侧将形成浓度尖峰,而相关计算显示该峰宽度小于相邻原子间距,这在物理上是难以接受的。合金元素浓度尖峰的存在仍有很大争议[6,15],Guo等[89]利用STEM-EDX分析发现,随着等温时间的延长,界面处Mn的浓度峰会发生数十纳米的扩展。EELS虽然可实现小于1 nm精细尺度的测量,但却鲜有对界面处Mn含量精确测量的实验报道[90,91]。近些年,3DAP技术的发展为探测原子空间级的元素分布提供了可能[92,93],可得到原子尺度的合金元素浓度分布。Danoix等[92]利用3DAP和STEM-EDX测得Fe-C-Mn合金α/γ界面附近C及Mn含量分布,发现C、Mn在界面附近存在显著浓度富集。
图3 不同浓度Fe-Ni、Fe-Mn和Fe-Co合金中奥氏体-铁素体相变开始温度Fs和铁素体-奥氏体相变开始温度As与新相和母相自由能相等温度(T0)的偏差[
Fig.3 Deviations of Fs and As from T0 upon cooling and heating in the Fe-Ni, Fe-Mn and Fe-Co alloys as a function of the solute concentration[
图4 Fe-Ni、Fe-Mn和Fe-Co合金中界面迁移率和温度的关系[
Fig.4 Derived interface mobility (Mint) as a function of temperature[
在传统的实验手段下,界面迁移不可避免地受到新相形核、晶体学与观察面取向关系以及相变后期软碰撞效应的影响,难以有效地探究元素配分行为对界面迁移的作用。近年来,随着对奥氏体/铁素体界面迁移与元素配分研究的不断深入,涌现出了几种新型研究方法。
2.2.1 梯度实验 Hutchinson等[94]将Fe-1Ni与Fe-5Ni (质量分数,%)合金用气动压力机在高温下压合,并在高温形成扩散偶,使试样中Ni含量呈梯度分布。其后通入CO2/CO混合气流进行渗碳处理,并调整混合气体比例实现基体C含量的控制,再对得到的梯度材料试样进行铁素体相变。金相组织观察结果表明Fe-0.1C-xNi (质量分数,%)合金相变动力学转变约发生在3.5%Ni (质量分数)附近,高于LE模型预测的2.9%Ni (质量分数)。梯度实验为研究奥氏体-铁素体相变动力学转变提供了高通量的方法,同时也可进一步拓展至晶粒长大、析出等具有类似转变行为的物理过程研究中。
2.2.2 脱碳实验 Zurob等[95,96,97]提出了等温脱碳的实验方法,将样品置于含少量水蒸气的氢气中,使其氧分压足以让样品表层脱碳,而不至于氧化,随后淬火至室温测量铁素体层的厚度。此方法排除了形核以及相变后期奥氏体内C扩散场软碰撞效应对界面条件的影响,同时可以在宏观尺度上对铁素体层厚度进行较为精确的测量,从而有效地对铁素体相变动力学进行定量研究。
在Fe-C-M (M=Mn、Ni、Cr、Mo等)体系的脱碳实验研究[95,96,97,98,99,100]中发现,含有Ni/Mn的三元合金在大部分实验温度内的铁素体相变动力学与LE模型吻合较好,其中含Mn的三元合金在高温时的铁素体相变动力学与PE模型吻合,随温度降低则逐渐向LE模型预测结果靠近,这一现象可归结为溶质原子的浓度尖峰需一定时间随界面迁移逐渐形成。与此相对应的是含Cr/Mo的三元合金中铁素体长大明显慢于PE和LE模型预测的长大速率,这是因为这些元素通常具有较强的界面偏聚倾向,可能对界面迁移形成额外的阻力,引起较强的溶质拖曳效应。
2.2.3 循环相变实验 Chen等[7,101]提出了一种循环相变方法对铁素体相变进行研究,即在奥氏体、铁素体、渗碳体平衡共存温度A1与奥氏体中开始析出铁素体的平衡温度A3之间选取2个特征温度,并在其间循环往复进行奥氏体-铁素体和铁素体-奥氏体相变,最终将试样冷却至室温。借助高温原位激光共聚焦显微镜观察,在循环的过程中没有观察到新铁素体晶粒的形核,且γ /α界面基本沿原路径往复迁移[102]。该方法的优点在于可以排除复杂的形核过程对相变动力学的影响,实验结果可以直接反应相变过程中界面迁移动力学。
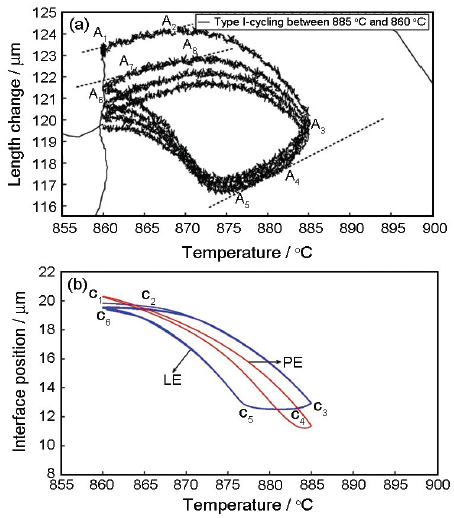
图5 Fe-0.17Mn-0.023C (质量分数,%)合金在885~860 ℃间进行γ-α循环相变中膨胀量随温度的变化及PE和LE模型对α/γ界面位置随温度变化的模拟结果[
Fig.5 Dilation as a function of temperature during cyclic experiments (a) and α/γ interface position as a function of temperature simulated under both local equilibrium and paraequilibrium conditions between 885 and 860 ℃ in Fe-0.17Mn-0.023C (mass fraction, %) alloy (b)[
循环相变研究发现了3种新的现象:(1) 转变停滞期[101],即温度发生改变,而相变量不变,如图5a[101]所示。停滞温度区间大小直接取决于合金元素种类与含量[103];(2) 逆转变期[101],即界面迁移方向与温度改变方向相反;(3) 长大滞缓期[104],即在最后冷却过程中,当温度降低到循环的初始温度附近时,界面移动突然减速,如图6a[104]所示。研究发现,逆转变期是系统处于非平衡态所导致的,而转变停滞期和长大滞缓期是Mn在界面处的扩散导致的。其中长大滞缓现象首次从实验上间接地证明了相变过程中界面处存在Mn的浓度尖峰。同时,如图5b和6b所示,LE模型能够定性地描述循环相变中出现的上述现象,而PE模型计算结果与实验不符。
图6 Fe-0.49Mn-0.1C (质量分数,%)合金在842~785 ℃间进行γ-α循环相变中膨胀量随温度的变化及经过1、2、6个循环后Mn的浓度分布模拟结果[
Fig.6 Dilations as a function of temperature during the γ-α cyclic phase transformations with 6 temperature cycles between 842 and 785 ℃ in a Fe-0.49Mn-0.1C (mass fraction, %) alloy (a) and simulated residual Mn spikes in austenite after cyclic phase transformations with 1, 2 and 6 temperature cycles (b)[
经过过去几十年的努力,奥氏体-铁素体相变过程中的界面迁移和元素配分研究取得了长足的进展,但是仍然存在诸多尚未解决的问题,可以预见这些依然将是未来的研究热点和难点。
在现有基于溶质拖曳理论的混合生长模型中,相变过程中界面处浓度尖峰的形成均是基于稳态假设[31,32,33,34,35],即假设浓度尖峰是瞬间形成的。然而,浓度尖峰的形成需要一定的时间,其与元素的扩散系数(包括界面内和奥氏体或铁素体相中的扩散系数)、界面处驱动力等热力学/动力学因素相关,必然存在从非稳态到稳态的过渡,对这一动力学过程的深入研究将有利于进一步了解元素在界面附近的动态配分机制。元素在移动界面处动态配分行为是一个多尺度问题,随着第一性原理、分子动力学、晶体相场、相场等多尺度计算模型的发展,可以预期人们对该问题的认识将会更加深入。
虽然文献中对三元合金Fe-C-M中奥氏体-铁素体相变进行了大量的研究,但是C和合金元素M之间的相互作用对界面迁移与元素配分行为的影响在绝大多数理论模型中并未予以考虑。Danoix等[92]利用3DAP技术观测到Fe-C-Mn合金中C会促进Mn在界面处偏聚,二者存在共偏聚效应。与Mn相比,碳化物形成元素(如Cr、Mo、V、Nb等)与C在界面处的相互作用更加复杂。一方面,碳化物形成元素与C原子之间存在强烈的吸引作用且容易在界面处富集,会产生明显的溶质拖曳效应使界面迁移速率降低;另一方面,当碳化物形成元素在界面处富集达到一定程度时会与C结合形成碳化物,从而减弱溶质拖曳效应。同时,新形成的碳化物也将对界面迁移起到一定的钉扎作用。
对于四元以及更多组元合金,不仅存在C和合金元素间的相互作用,不同合金元素之间也可能会在界面处发生相互作用。Sun等[105]对Fe-C-Mn-Mo四元合金中的耦合溶质拖曳效应进行了脱碳实验研究,发现理论上将Fe-C-Mn与Fe-C-Mo三元合金的溶质拖曳效应叠加可以很好地解释Fe-C-Mn-Mo四元合金的实验相变动力学。然而,Qiu等[44]却发现若同样地将Mn与Si对应三元合金的溶质拖曳效应叠加,将会高估Fe-C-Mn-Si四元合金中的溶质拖曳效应程度。考虑到C-Si与C-Mo之间存在着截然相反的相互作用,C对合金元素-界面相互作用的影响被认为可能是造成理论与实验偏离的主要因素。然而这一推论有待在无C的合金系中进行实验验证,从而排除Mn-Si和Mn-Mo合金元素之间的相互作用影响。总体来说,钢中不同元素之间的相互作用机理及其对奥氏体-铁素体相变动力学的影响非常复杂,需进行深入的实验和理论研究。
大多数现有相变模型对相界面进行了较大程度的数学简化与近似,相关的界面物理量(界面厚度、界面内扩散系数、元素-界面结合能等)也大多作为拟合参数,导致模型的预测能力有限。未来原子尺度计算模型(如第一性原理、分子动力学等)以及原子尺度实验表征技术(如三维原子探针、高分辨透射电镜等)的发展,将获得更加准确的界面结构信息以及界面相关的物理参量,指导相变理论模型的发展。
The authors have declared that no competing interests exist.

/
| 〈 |
|
〉 |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}